Unknown proteins in Mycobacterium tuberculosis
Background
Genome annotation is the process of locating and describing the function of all relevant sequence regions in the genome. The simplest way to perform gene annotation is to identify open reading frames for coding genes (ORFs) and then use homology based search tools, like BLAST, to search for homologous genes. Annotation is now largely automated for the bulk of new genomes because there is so much new sequence being produced and much of it can be assigned function based on known information. However in the early days, the relatively few bacterial genomes would be manually curated. The reference genomes created then still exist, some as ‘gold standard’ annotations that have been maintained ever since. Some older annotations are less well maintained and perhaps have been superseded. These still appear on GenBank as reference genomes and you can see how this might confuse potential users as to which is the correct annotation to use.
Update
This paper now details the updates discussed here to the M.bovis annotation.
Mtb H37Rv
H37Rv is a widely used laboratory strain of Mycobacterium tuberculosis (Mtb) and was the first Mtb genome to be annotated in 1998 by Stewart Cole and collegues. (Another assembly was made by the Broad institute and six strains of H37Rv from distinct laboratories were sequenced in 2010 by Texas AM researchers). The original annotation has been updated and curated since and is still considered the reference genome for this strain. This annotation was long made accessible through the Tuberculist database and now Mycobrowser.
Hypothetical proteins
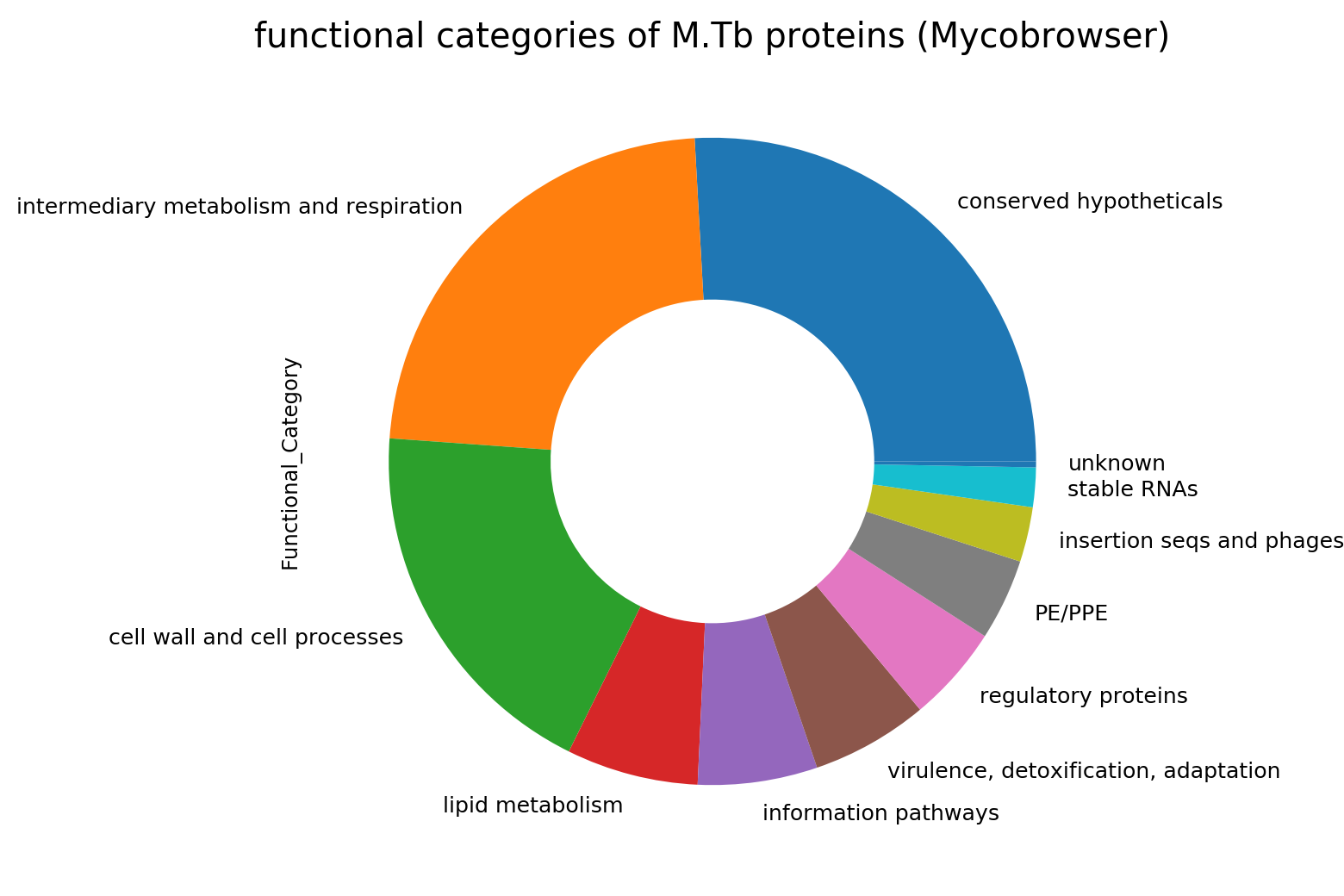
In the initial annotation, the function of about one-quarter of the Mtb coding genome was unknown. When proteins but can’t be identifed as to function they have been traditionally assigned names like ‘unknown protein’, ‘hypothetical protein’ or ‘conserved hypothetical’. Those familiar with the Mtb genome may recognise the functional categories as originally assigned shown in the pie chart at right. Conserved hypotheticals make up a substantial number. Interestingly, the other annotations have assigned more proteins function and have less hypothethicals as seen in the table below. Sanger refers to the reference genome. This indicates that the original annotation is somewhat outdated in this regard.
| genome | coding sequences | cds with gene names | hypothetical |
|---|---|---|---|
| sanger | 4018 | 1953 | 1097 |
| broad | 4143 | 16 | 806 |
| siena | 4152 | 317 | 799 |
| LPb | 4111 | 16 | 809 |
Later sources of annotation
More recently the PATRIC database have used their own pipeline based on RASTtk to re-annotate H37Rv which diverges from the reference due to some additional functional assignments and additional small genes that are not in the reference.
Doerks at al. in 2012 added approximately 620 new functional assignments to the remaining unknowns in H37Rv. This was done using orthology and genomic context evidence. If a hypothetical protein was a member of a known orthologous group in the eggNOG database, this annotation was brought over to the Mtb protein. The STRING tool was also used, combining gene fusion events and significant co-occurrence to predict links with known proteins. The annotations vary in specificity. Though in some cases they are likely very accurate predictions, others could be seen as ‘functional hints’ of the nature of the protein. Where these new additions fall in the Tuberculist functional categories is shown below:
Some of the genes found by Doerks et al. are also assigned in PATRIC and a few have since been assigned in the reference. We can see that there is an overlap in each set with the old reference in the venn diagram below of all existing unknown proteins. This shows that if all the annotations were to be combined we would be only left with about 400 strictly hypothetical proteins in the genome (the brown region in the middle):
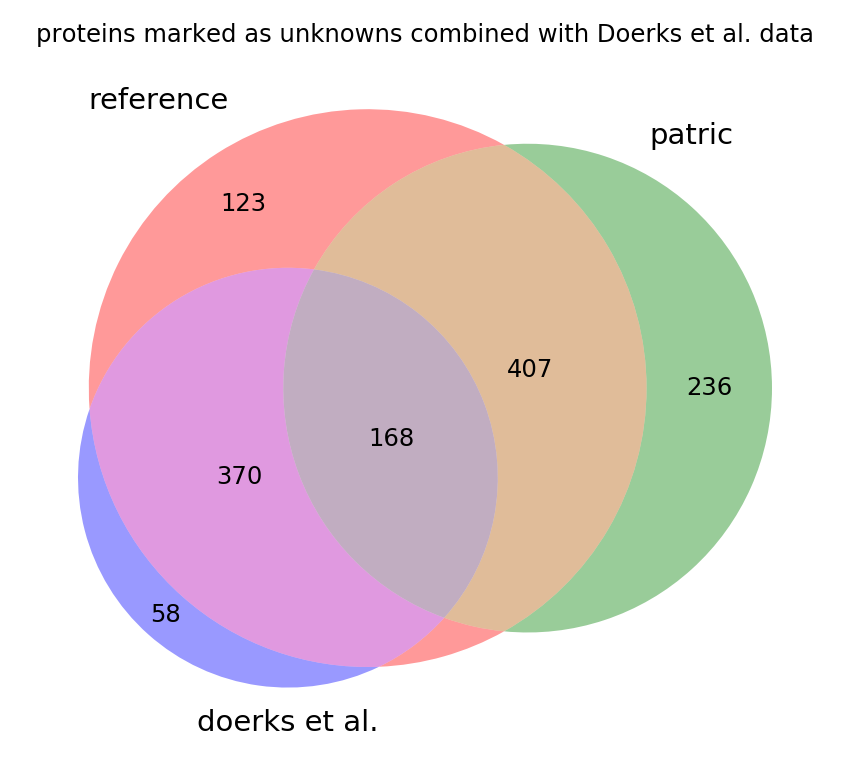
This information is yet to be integrated into the reference annotation.
Structure based annotation
Finally, Anand at al. (2011) have used structure based prediction methods to predict the function of multiple hypotheticals in the Mtb proteome. Molecular models were obtained from MODBASE and used to identify function in different ways. First through known fold-function associations, second through identification of known sub-structural motifs and third through binding site identification. One example is Rv1752, which they predict to be an oxidoreductase whose co-enzyme binding requirements are met by its gene-neighbor Rv1751, a potential FAD-binding monoxygenase. Thus non-sequence based methods are a valuable tool in the annotation process and probably under utilised.
Which annotation to use?
Clearly an advantange of tools like PATRIC is the standardization of annotations across genomes. It has an integrated pipeline that can keep annotations up to date and useful interactive tools. However it is still a specialist database and not universally known. It is not going to replace the use of GenBank/EBI. Users going to these data sources may not distinguish between annotations or even assemblies correctly. The reference is often used by default. Many papers cite the the original H37Rv genome annotation and it will remain used by TB researchers for the foreseable future. It is therefore important to keep these annotations up to date. The annotations discussed here have been combined together into a single table that may prove useful to TB researchers. It is linked below. This includes the structural/fold predictions from Anand et al.
Links
- Table with annotations combined
- H37Rv genome on genbank
- Broad H37Rv genome
- Mycobrowser
- PATRIC M.tb (broad)
- RAST tool kit
- Prokaryotic RefSeq Genomes FAQ
References
- Thomas R. Ioerger et al. Variation among Genome Sequences of H37Rv Strains of Mycobacterium tuberculosis from Multiple Laboratories. Journal of Bacteriology Jun 2010, 192 (14) 3645-3653; DOI: 10.1128/JB.00166-10
- Joseph J. Gillespie et al. PATRIC: the Comprehensive Bacterial Bioinformatics Resource with a Focus on Human Pathogenic Species. Infection and Immunity Oct 2011, 79 (11) 4286-4298; DOI: 10.1128/IAI.00207-11
- Doerks T, van Noort V, Minguez P, Bork P (2012) Annotation of the M. tuberculosis Hypothetical Orfeome: Adding Functional Information to More than Half of the Uncharacterized Proteins. PLOS ONE 7(4): e34302. https://doi.org/10.1371/journal.pone.0034302
- Anand P, Sankaran S, Mukherjee S, Yeturu K, Laskowski R, et al. (2011) Structural Annotation of Mycobacterium tuberculosis Proteome. PLOS ONE 6(10): e27044. https://doi.org/10.1371/journal.pone.0027044
- Mitra P, Shultis D, Brender JR, Czajka J, Marsh D, et al. (2013) An Evolution-Based Approach to De Novo Protein Design and Case Study on Mycobacterium tuberculosis. PLOS Computational Biology 9(10): e1003298. https://doi.org/10.1371/journal.pcbi.1003298