A whole genome MLST (wgMLST) implementation in Python
Background
MLST (Multi Locus Sequene Typing) is a well known bacterial genotyping method with applications in pathogen surveillance and phylogenetics. It means selecting a handful of well conserved (or ‘housekeeping’) genes and comparing their sequences across samples. The alleles of each sequence are coded are stored in a reference database for comparison. wgMLST (or cgMLST) is an extension of the traditional method to many genes, enabled by the use of whole genome sequencing technology. The idea is to identify a set of stable genes in a well annotated genome and identify all the alleles in that sample from sequence read data. This is a much higher resolution version of the traditional method. In this way isolates can be compared consistently across labs. For non-clonal species with a lot of genome variation this means the ‘core genome’ for the species is used. For very clonal populations like members of the Mycobacterium TB complex (MTBC) many genes will always be present reliably and can be included. This has been well described by Kohl et al.
The other approach typically used in bacterial epidemiology of whole genome data is the ‘map to reference and SNP site analysis method’. This works best for highly clonal populations and is probably less effective in other species. One other reason for applying wgMLST instead is really for standardization purposes. It’s not really possible to establish a standard set of SNPs across a population, as there will always be unknown SNP sites in as yet unseen samples (though a set representative set of sites might be sufficient). Some resolution is sacrificed with wgMLST compared to SNP analysis. Also it can’t be used for phylogenetic inference.
Studies implementing this method of MLST on sequence data mainly to use commercial solutions such as BioNumerics or Ridom SeqSphere+. The huge downside to this is that any lab wanting to use the same system has to have access to this software or at least to the reference database of alleles that has to be shared across labs. It is possible that these databases can be made public and another piece of software could contribute to them in parallel, but I have not seen evidence that this is the case. In effect these proprietary solutions are not yet compatible with open standards. Here is described a wgMLST method implemented in Python as part of the open source pathogenie package. It is designed to be used for M.bovis but can be applied to other species.
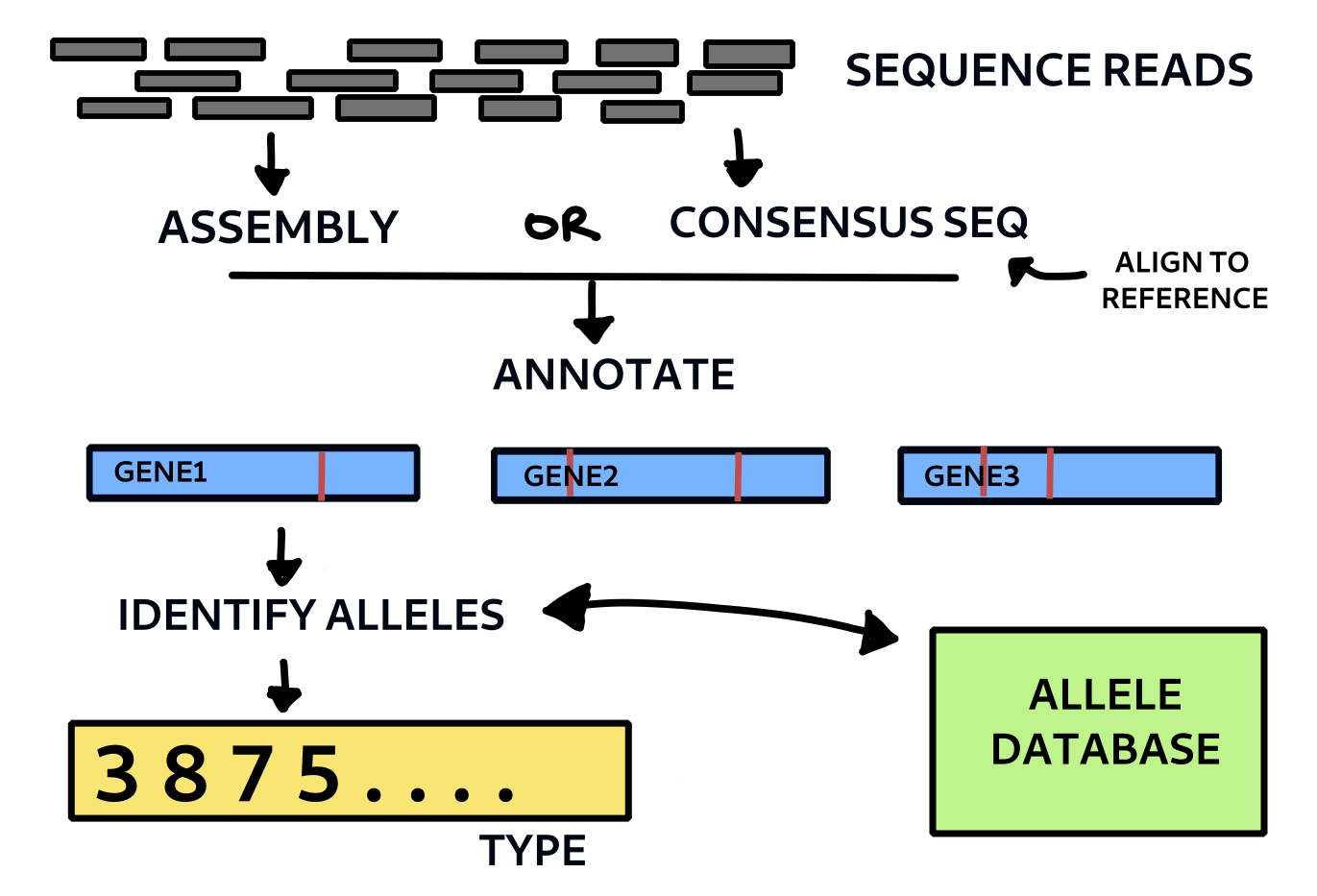
Basic outline of wgMLST method. Each allele is given a code number and each is checked against a database of known allelic variants for that species.
MLST scheme
This is the set of genes chosen to serve as the core gene set for comparison. The 2891 genes used by Kohl et al. for the Mycobacterium TB lineage are provided at cgmlst.org. These genes were used here to create a M.bovis specific list with the locus tag as the identifier and the strain AF2122/97 as the reference genome. This list was used to match sequences from the reference genome annotation. It serves as the basis for making our initial allele table. Every time we identify a new allele, it is added to this table by incrementing the number. So the information stored per allele is as follows:
name allele sequence
Mb0014c 1 CTACTGGCCGAACCTCAGCGTGATGATGC...
Mb0015c 1 TCATTGCGCTATCTCGTATCGGGCCAGCG...
Mb0016c 1 TCATGGTTCCCCCTGCAGTGCGGCTTCGA...
Mb0017c 1 TCATACGCGTTCGATGACCTCGGTGCCGT...
Code
The entire code can be found in the wgmlst Python module in the pathogenie github repository here. Below are just given some of the essential functions and outline for running the analysis. The remaining functions are all in the module. type_sample takes a fasta file of the genome sequence as input. This could come from assembling the reads or finding the consensus sequence from an alignment. In the case of M.bovis the latter might be sufficient and will capture SNPs and most indels. (It will miss large insertions not present in the reference). For other species assembly is probably needed. The method here uses consensus sequences derived from the multi sample vcf file of a previous variant calling run with SNiPgenie (another pipeline could be used too). It then annotates the sequence using the run_annotation and returns a set of records that are converted into gene by gene nucleotide sequences. The method find_alleles (not shown here) then goes through each sequence and checks it against the known ones (from a database or flat file). If they match the code is applied, otherwise a new entry is created and a code number assigned to the new allele. Thus the importance of updating and sharing the allele database is clear for standardisation of results. The result for each sample is a set of profiles with a number for each allele that can be compared together by building a distance matrix. This can then be used to make a neighbour joining or minimum spanning tree, for example.
def type_sample(fastafile, outfile, threads=4, overwrite=False):
"""Type a single sample using wgMLST.
Args:
fastafile: fasta file to type from assembly or other
path: output folder for annotations
Returns:
dataframe of MLST profile
"""
if overwrite == True or not os.path.exists(outfile):
#annotate
featdf,recs = run_annotation(fastafile, threads=threads,
kingdom='bacteria', trusted=ref_proteins)
#get nucl sequences from annotation
SeqIO.write(recs,'temp.gb','genbank')
get_nucleotide_sequences('temp.gb',outfile,idkey='protein_id')
#find alleles
res,new = find_alleles(outfile)
#update db
update_mlst_db(new)
return res
def run_samples(vcf_file, outdir, omit=[], **kwargs):
"""Run samples in a vcf file.
Args:
vcf_file: multi sample variant file from previous calling
outdir: folder for writing intermediate files
Returns:
dict of mst profiles
"""
profs = {}
samplenames = get_samples_vcf(vcf_file)
for s in samplenames:
if s in omit:
continue
get_consensus(vcf_file, s)
outfile = os.path.join(outdir, '%s.fa' %s)
profile = type_sample('consensus.fa', outfile, **kwargs)
profs[s] = get_profile_string(profile)
return profs
profs = run_samples(vcf, outfolder)
D = dist_matrix(profs)
tree = tree_from_distmatrix(D)
tree.write('mlst.newick', 'newick')
To run this from the command line you can install pathogenie with pip use the pathogenie-wgmlst command. It currently only works for M.bovis. The mlst database is stored as a csv file in this case but this could be updated to allow use of a database such as mysqlite that can be
pathogenie-wgmlst -i <input vcf> -o <output folder for annotations> -t <threads>
Testing
This code is tested a follow up post.