An MHC-Class I binding predictor with sklearn, part 2
Background
This post follows on from a previous one about making an MHC-I binding predictor using scikit-learn in Python. The technique is detailed fully there. To improve the predictor we made before it was only necessary to use the GridSearchCV method of sklearn to search over an appropriate parameter space for the MLPRegressor. This is an implementation of a multilayer perceptron (MLP), a class of artificial neural network.
Code
To run the grid search we use this code. This takes some time as it runs training for each combination of the parameters which can be hundreds of times.
from sklearn.model_selection import GridSearchCV
from sklearn.neural_network import MLPRegressor
import epitopepredict as ep
df = ep.get_training_set('HLA-A*03:01', length=9)
X = df.peptide.apply(lambda x: pd.Series(blosum_encode(x)),1)
y = df.log50k
param_list = {"hidden_layer_sizes": [(1,),(50,)],
"activation": ["identity", "logistic"], "max_iter": [200,1500],
"solver": ["adam", "lbfgs", "sgd"], "alpha": [0.00005,0.0005]}
reg = MLPRegressor()
gs = GridSearchCV(reg, param_grid=param_list)
gs.fit(X, y)
gs.best_params_
You could also used a Random search through the parameter space using RandomizedSearchCV.
from sklearn.model_selection import RandomizedSearchCV
distributions = dict(hidden_layer_sizes=np.random.randint(20,200,10),
activation=["identity", "logistic"],
alpha=np.random.uniform(.00001,0.001,10),
solver=["adam", "lbfgs"])
reg = MLPRegressor()
rs = RandomizedSearchCV(reg, distributions, random_state=2, n_iter=20)
search = rs.fit(X, y)
search.best_params_
The encoder
The peptide encoder for representing sequences of amino acids has been detailed previously. These encoding create a vector of numbers and is what we fit to the target values (in this case binding affinities). Testing this further shows little difference between the one hot encoding and blosum encoding methods as below. The AUC is the average of a five fold cross validation.
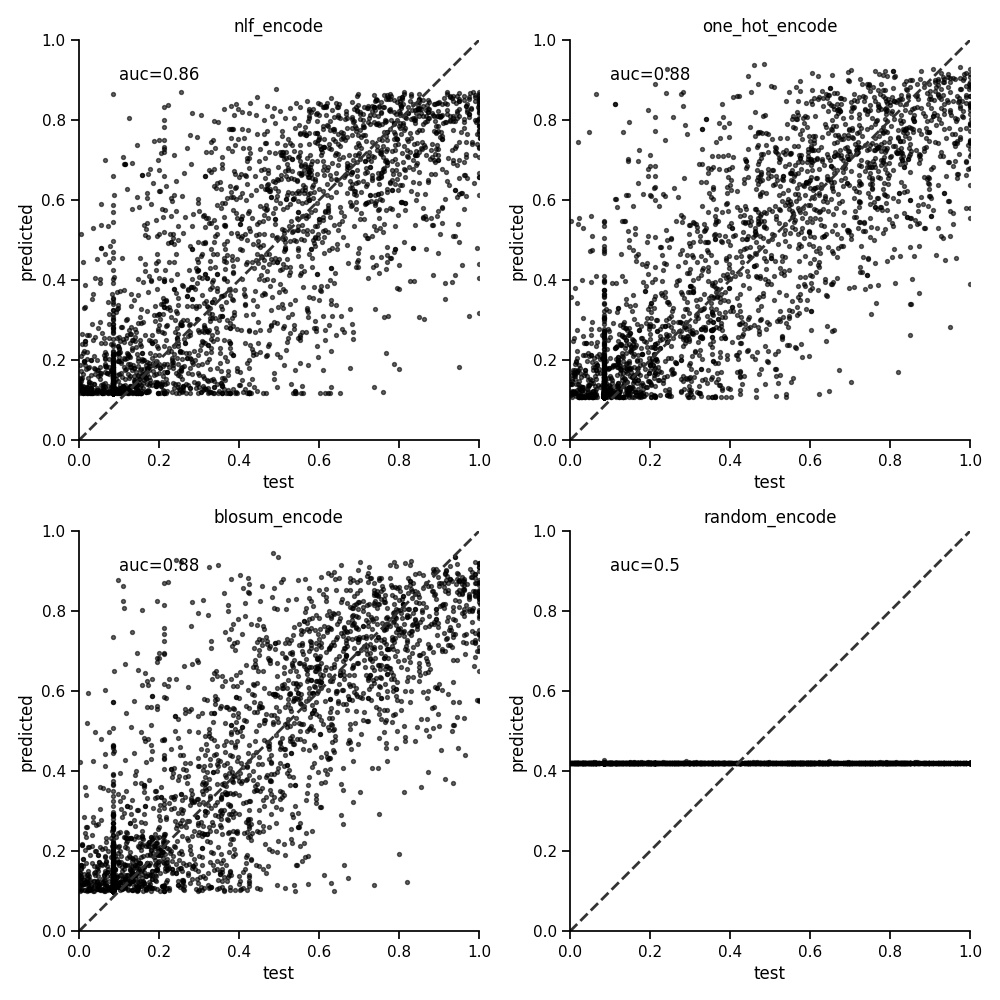
Running for a selection of alleles with different sample sizes shows a similar result. We therefore use the one hot encoder below.
| encoder | HLA-A*01:01 | HLA-A*02:01 | HLA-A*03:01 | HLA-A*24:02 | HLA-B*07:02 | HLA-B*44:03 | mean |
|---|---|---|---|---|---|---|---|
| nlf | 0.829 | 0.867 | 0.833 | 0.751 | 0.860 | 0.746 | 0.814 |
| one hot | 0.851 | 0.880 | 0.835 | 0.740 | 0.872 | 0.763 | 0.824 |
| blosum | 0.842 | 0.879 | 0.839 | 0.752 | 0.866 | 0.761 | 0.823 |
| random | 0.500 | 0.501 | 0.500 | 0.498 | 0.503 | 0.500 | 0.500 |
Comparison to other tools
The two best MHC-class I prediction tools are currently netMHC/netMHCpan and MHCFlurry. Here we compare our predictor using an evalutation set not used for training. We used the training set compiled by the authors of MHCFlurry. The evaulation peptides are from Kim et al (2014) and available from the IEDB.
For this test netMHCpan 4.1b and MHCFlurry 2.0.1 were used. The auc is used as the metric with 500 ic50 value as the threshold.
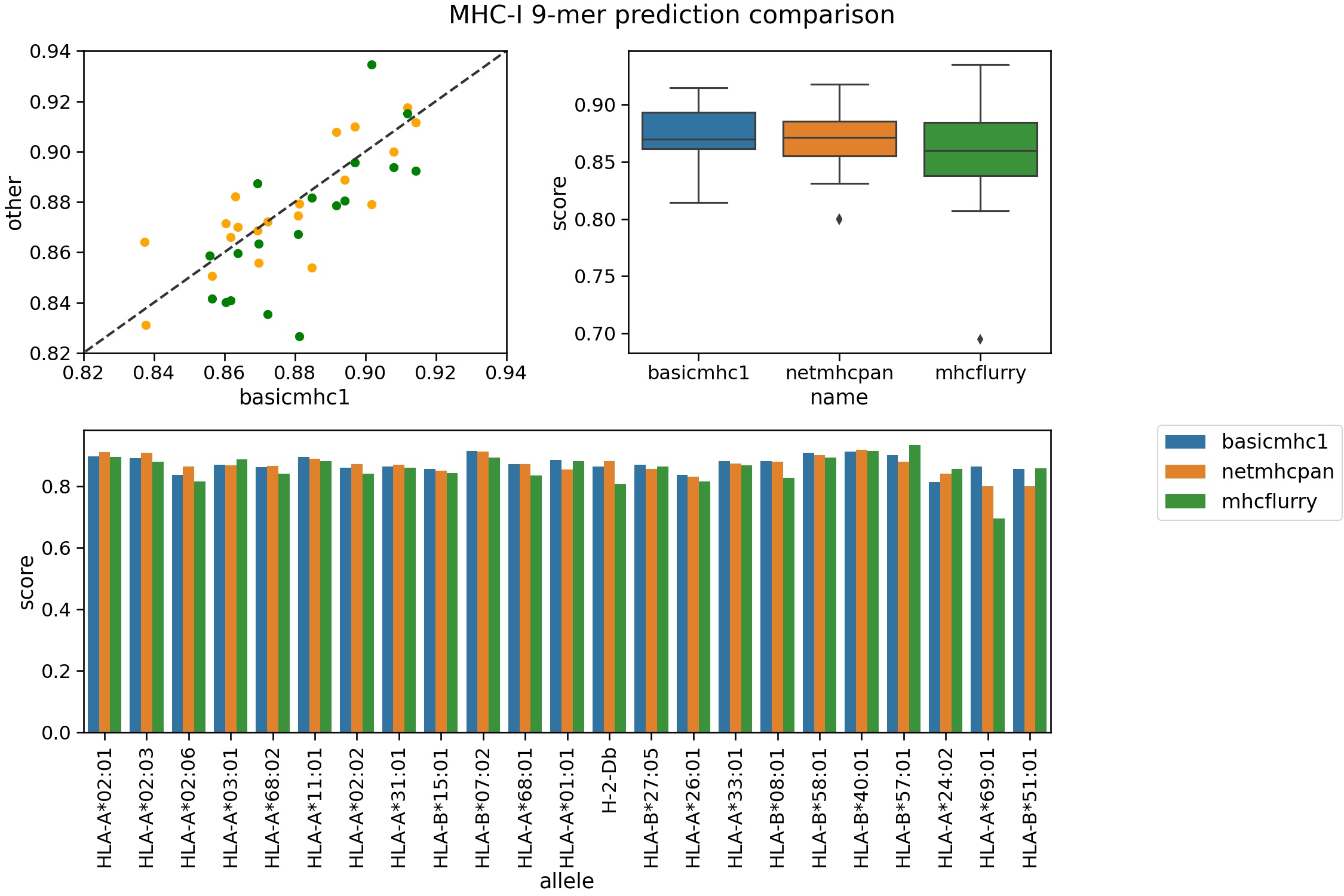
Rigorous benchmarking is something of a technical skill in itself. This should be seen as a rough guide of the performance of the tools. It shows our method is in the ballpark of the other programs. The other two are ‘pan specific’ predictors in that they can be applied to other alleles without training data. Ours is not and thus is more limited in scope. netMHCpan 4.1 can predict many alleles over a broader length range. Only binding affinities are predicted here (not ligand likelihood).
Overlap of predicted bnders in a protein
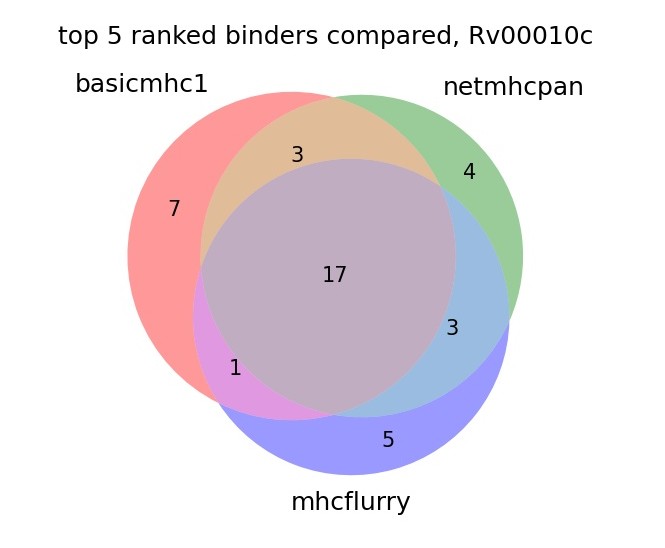
To check our model with more real use data we can predict the binders in a protein sequence and compare the top ranked from each method. Here we use the protein sequence from Mtb, Rv00010c. The plot shows the overlap of the top 5 ranked from each method in 6 alleles.
epitopepredict
This method is implemented in the epitopepredict package as the built-in MHC-I predictor. You can use it as follows:
import epitopepredict as ep
from epitopepredict import base, mhclearn
mhclearn.train_models()
m1_alleles = ep.get_preset_alleles('mhc1_supertypes')
seq = 'MTDDPGSGFTTVWNAVVSELNGDPKVDDGP'
P = base.get_predictor('basicmhc1')
b = P.predict_sequences(seq, alleles=m1_alleles, length=9, cpus=8)
Links
References
- Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: Improved Peptide–MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J Immunol. 2017; doi: 10.4049/jimmunol.1700893.
- T. J. O’Donnell, A. Rubinsteyn, M. Bonsack, A. B. Riemer, U. Laserson, and J. Hammerbacher, “MHCflurry: Open-Source Class I MHC Binding Affinity Prediction,” Cell Syst., vol. 7, no. 1, p. 129–132.e4, 2018.
- https://www.iedb.org/