Calculate PubMLST sequence types using Python
Background

PubMLST, or Public databases for Molecular Typing and Microbial Genome Diversity, is a web-based platform that provides access to databases of microbial genetic sequences. It is primarily used for microbial typing, strain comparison, and epidemiological studies. The primary purpose of PubMLST is to facilitate the standardization and sharing of microbial sequence data. The databases hosted by PubMLST typically include sequence types (STs), allelic profiles, and sometimes additional metadata for each strain. Users can query the databases, submit their own sequences for analysis, and access tools for phylogenetic analysis, sequence alignment, and other analyses relevant to microbial genomics. Each species has it’s own database.
If you have whole genome sequences you can use pubMLST data to calculate sequence types locally. You might want to do this to avoid having to upload many sequences to the database. This is accomplished below using some Python code. The species Mycoplasma bovis is used in the example but it should work for any species database.
ST and allelic profile
Briefly MLST works by recording alleles for specific set of conserved loci in the genome. Typically 6 or 7 genes/loci are used. Each species has it’s own custom set of loci. Each allele is given a number in order of discovery and so the allelic profile is just a set of numbers allocated to a strain for each locus. This profile is a proxy for genetic relatedness. PubMLST databases then keep a record of strains and their types, allelic profile.
For the example of Mycoplasma bovis there are 7 loci in the scheme, developed by Karen Register (USDA). You can get an idea of what the profiles look like below:
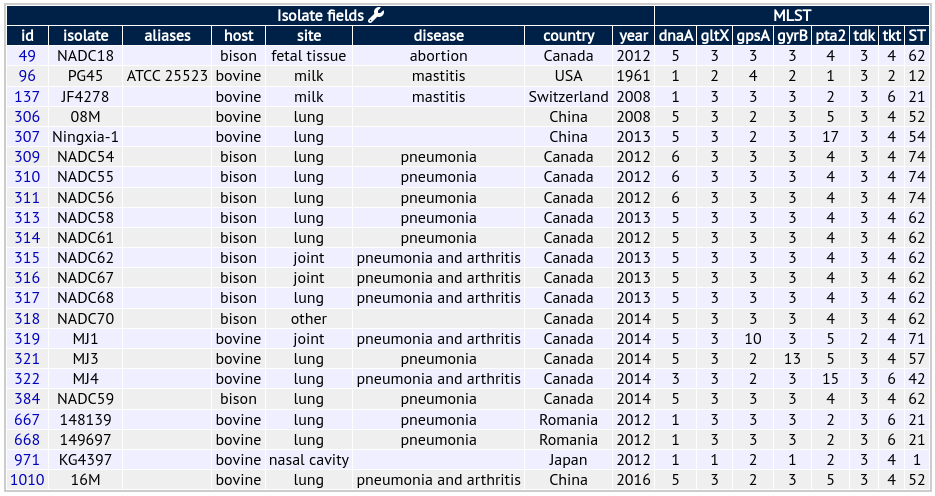
Retrieving profiles and known sequences
PubMLST has a REST interface for web requests. It can be used to download the profiles of already known strains and the sequences for each locus with all the known alleles and ST number.
Retrieve gene sequences of all alleles for each locus at:
https://rest.pubmlst.org/db/pubmlst_mbovis_seqdef/loci/{locus}/alleles_fasta
Retrieve existing profiles:
https://rest.pubmlst.org/db/pubmlst_mbovis_seqdef/schemes/2/profiles_csv
Code
This code just takes the allele sequences of each locus and blasts the assembly to it. Exact matches to an allele are recored. If there’s no hit that allele is not in the database. We then just decode the numbers into an ST using the profiles data make_blast_database and blast_fasta are utility functions in the tools module of the SNiPgenie package. You can download the code for those methods from here or install the package and import them as below.
Note the use of a cutoff option for the percentage identity from the blast. This should really be 100% since we want exact hits to each allele. However if you wanted to allow for errors in the query sequence this could be lowered to 99.5 perhaps. This could create false positive hits however.
import os
import pandas as pd
from snipgenie import tools
#loci defined specfic to organism
loci = ['dnaA','gltX','gpsA','gyrB','pta2','tdk','tkt']
for gene in loci:
db = f'MLST/{gene}.fa'
tools.make_blast_database(db)
def get_pubmlst_profile(fasta_file, cutoff=100):
"""try to get pubmlst profile for an assembly"""
profile=[]
for gene in loci:
db = f'MLST/{gene}.fa'
bl = tools.blast_fasta(db, fasta_file)
bl = bl[(bl.pident>=cutoff)]
if len(bl)>0:
#get top result if more than one
res = bl.iloc[0]
hit = res.stitle
else:
hit = None
profile.append(hit)
#extract ST numbers from locus names
profile = [i[-1:] if i!=None else None for i in profile]
return profile
def get_st(profile):
"""Get ST from profile"""
df = pd.read_csv('MLST/profiles.csv',sep='\t').astype(str)
if None in profile:
return
s = ''.join(profile)
#iterate over profiles to find a match
for index, row in df.iterrows():
row_str = ''.join(list(row[1:8]))
#compare the profile strings
if s == row_str:
return row.ST
return
We can then use the functions to run sequences in a batch. The code below loops over a list of files and adds the results to a dataframe.
res=[]
paths = glob.glob('assembly/*.fa') #path to your fasta files
for file in paths:
name = os.path.splitext(os.path.basename(file))[0]
p = get_pubmlst_profile(file)
st = get_st(p)
#print(file,p,st)
res.append([name,p,st])
res=pd.DataFrame(res,columns=['name','profile','ST'])
res.to_csv('st_types.csv',index=False)
Links
References
- Register KB, Lysnyansky I, Jelinski MD, Boatwright WD, Waldner M, Bayles DO, Pilo P, Alt DP. Comparison of Two Multilocus Sequence Typing Schemes for Mycoplasma bovis and Revision of the PubMLST Reference Method. J Clin Microbiol. 2020 May 26;58(6):e00283-20. doi: 10.1128/JCM.00283-20. PMID: 32295891; PMCID: PMC7269390.