Bacterial species identification from WGS using 16S genes
Background

The 16S rRNA gene is present in all bacteria and contains both highly conserved and hypervariable regions. The conserved regions allow for the design of universal primers that can amplify the gene across a wide range of bacteria and archaea. The hypervariable regions (e.g., V1-V9) provide species-specific sequences that can be used to distinguish between different taxa. This is a widely used means of species identification. With WGS we have the whole genome and relying on 16S alone is not necessary. However it is a useful first step in checking your sequences are what you think. The only snag for this method is that you need to assemble the reads first.
Basically this method just takes the assembly and does a local blast to the 16S ncbi sequences. The results are then sorted by precent identity and coverage. You can download the 16S sequences from here. Note that the tools module from snipgenie is imported here for some of the functions.
import numpy as np
import pandas as pd
import pylab as plt
from Bio import AlignIO, SeqIO
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
from snipgenie import tools
def get_blast_coverage(bl, fasta):
"""Get alignment coverage of blast results from original sequence lengths"""
df=tools.fasta_to_dataframe('16S_ncbi.fa')
df=df.rename(columns={'length':'sslen'})
bl=bl.merge(df,left_on='sseqid',right_on='name',how='left')
bl['perc_cov'] = bl.apply(lambda x: round(x.length/x.sslen*100,2),1)
return bl
def blast_16S(filename, hits=100, pident=99.5):
"""Perform blast to 16S genes"""
tools.make_blast_database('16S_ncbi.fa')
bl=tools.blast_fasta('16S_ncbi.fa',filename,maxseqs=hits)
bl = get_blast_coverage(bl, '16S_ncbi.fa')
#bl['genus'] = bl.stitle.apply(lambda x: x.split()[1])
bl['species'] = bl.stitle.apply(lambda x: ' '.join(x.split()[1:3]))
cols = ['sseqid','sslen','length','perc_cov','pident','stitle','species']
bl = bl[(bl.pident>=pident) & (bl.perc_cov>=80)].sort_values('pident',ascending=False)
return bl
def extract_sequences_by_ids(input_fasta, output_fasta, ids_to_extract):
"""Extract sequences from fasta file with given ids"""
sequences = SeqIO.parse(input_fasta, "fasta")
# Filter sequences that match the given IDs
filtered = (seq for seq in sequences if seq.id in ids_to_extract)
SeqIO.write(filtered, output_fasta, "fasta")
return
def append_sequences_to_fasta(fasta_file, seqs):
"""Append SeqRecords to a FASTA file, overwriting the old file."""
existing_seqs = list(SeqIO.parse(fasta_file, "fasta"))
if type(seqs) is not list:
seqs = [seqs]
existing_seqs.extend(seqs)
#with open(fasta_file, "w") as output_handle:
SeqIO.write(existing_seqs, fasta_file, "fasta")
return
def get_tree(fasta_file):
"""Get phylo tree from fasta_file with fasttree"""
out = 'temp.newick'
cmd=f'mafft {fasta_file} > temp.aln'
tmp = subprocess.check_output(cmd, shell=True, stderr=subprocess.PIPE)
cmd=f'fasttree temp.aln > {out}'
tmp = subprocess.check_output(cmd, shell=True, stderr=subprocess.PIPE)
return out
Usage
Here is how the functions above are utilised to check an assembly. For this example I used an assembly of a known Mycoplasma alkalescens strain, selected at random. Note that you can just use the blast table to get the closest match. The other code is to make a phylogeny from the alignment which might give a less ambigiuous result for some cases.
#run local blast
tools.make_blast_database('16S_ncbi.fa')
bl = blast_16S('M.alkalescens_NCTC10135.fa',pident=90,hits=20)
#the following parts are if we want to make a tree
#add target sequence into results
targetseq = SeqRecord(Seq(bl.iloc[0].qseq), id='NCTC10135')
m=bl.set_index('sseqid')[:15]
names = list(m.index)
#extract the original sequences for our alignment
extract_sequences_by_ids('16S_ncbi.fa', 'temp.fa', names)
append_sequences_to_fasta('temp.fa',targetseq)
#get a phylogeny
treefile = get_tree('temp.fa')
Here’s the blast result table:
| sseqid | species | pident |
|---|---|---|
| NR_025984.1 | Metamycoplasma alkalescens | 99.932 |
| NR_026035.1 | Metamycoplasma auris | 98.973 |
| NR_025988.1 | Metamycoplasma canadense | 98.704 |
| NR_029180.1 | Metamycoplasma gateae | 98.545 |
| NR_041743.1 | Mycoplasmopsis arginini | 98.494 |
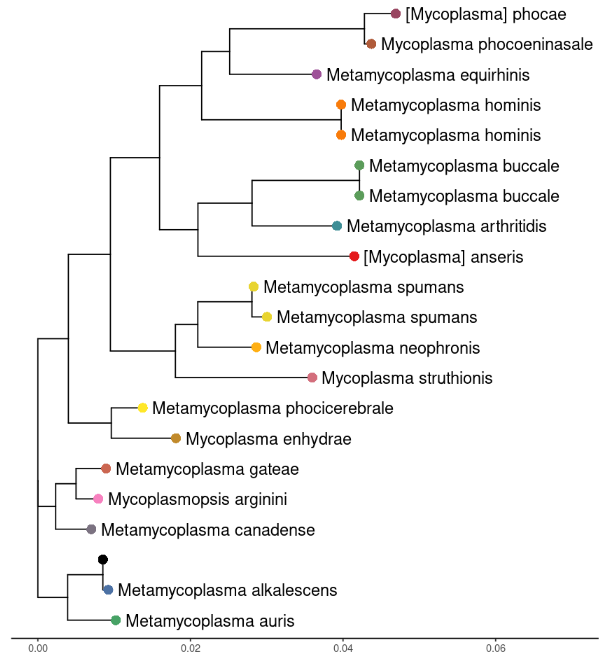
Here’s what the tree looks like with our assembly being the black tip. It is correctly identified as Mycoplasma alkalescens. Obviously results will vary depending on your sample. A mixed sample with DNA from two strains will need to be treated carefully.
There is a Jupyter notebook here with this code. This method is also implemented as plugin in the SNiPgenie GUI.
Links
- https://en.wikipedia.org/wiki/16S_ribosomal_RNA