Explore the SARS-CoV-2 spike protein sequences using Python tools
Background

Since the identification of SARS-CoV-2 virus and it’s subsequent spread, there has been considerable discussion and uncertainty over it’s origin. The genome of the newly emerging CoV consists of a single, positive-stranded RNA that is approximately 30k nucleotides long. The overall genome organization of the newly emerging CoV is similar to that of other coronaviruses. The newly sequenced virus genome encodes the open reading frames (ORFs) common to all betacoronaviruses, including the spike-surface glycoprotein (S). The S protein contains two functional domains: a receptor binding domain, and a second domain which contains sequences that mediate fusion of the viral and cell membranes. The S glycoprotein must be cleaved by cell proteases to enable exposure of the fusion sequences and hence is essential for cell entry. The receptor binding domain (RBD) in the spike protein is the most variable part of the virus genome. SARS-CoV-2 seems to have an RBD that may bind with high affinity to ACE2 from human and closely related mammals, but less so in other species. Six residues in the RBD appear to be critical for binding to the ACE2 receptor and determining host range. Five of these six residues are mutated in SARS-CoV-2 compared to the most closely related bat virus, RaTG13 (MN996532) (Anderson et al.). The sequence comparison also shows the insertion of a furin cleavage sequence in SARS-CoV-2. This may alter the ability of the virus to infect cells in humans relative to the Bat form. Interested readers should check the links at the end of this page for further details. www.virology.ws has some easy to read articles on current research.
This page illustrates some microbial genomics methods using Python by replicating some of the results dicussed by Andersen et al.. It is important to note that the content of this page is the subject of current and ongoing research and should not be seen as indicating a special expertise in virology on the part of the author.
The notebook with this code is available at this URL. This method will work for any set of bacterial/viral sequences you want to compare.
Imports
import os,sys,glob,random,subprocess
from importlib import reload
import pandas as pd
from Bio import SeqIO
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
from Bio import Phylo, AlignIO
import pathogenie
from pybioviz import plotters
from bokeh.io import show, output_notebook, output_file
output_notebook()
from ete3 import Tree, NodeStyle, TreeStyle, PhyloTree
Use the NCBI viruses database to download betacoronavirus sequences
NCBI Virus is a community portal for viral sequence data and it is easy to download nucleotide sequences for known betacoronavirus isolates here. This link links to all current sequences. Then we download the csv table and fasta file for these sequences. Subsets can then be used from this table, chosen by accession number or host name etc.
ncbidata = pd.read_csv('ncbi_betacoronavirus_25-02-20.csv')
print (len(ncbidata))
print (ncbidata.columns)
ncbidata['Release_Date'] = pd.to_datetime(ncbidata.Release_Date)
print (ncbidata.Host.value_counts()[:10])
#put sequences into a set of SeqRecord objects with Biopython
seqrecs = SeqIO.to_dict(SeqIO.parse('ncbi_betacoronavirus.fasta','fasta'))
Get a subset of the sequences
accessions = ['KY417146','MN908947','MN996532','AY278741','MK211376']
subset = ncbidata[ncbidata.Accession.isin(accessions)]
cols= ['Accession','Species','Host','Collection_Date']
subset[cols]
| Accession | Title | Host | Collection_Date |
|---|---|---|---|
| MK211376 | Coronavirus BtRs-BetaCoV/YN2018B | Rhinolophus affinis | 2016-09 |
| MN908947 | SARS-CoV-2 | Homo sapiens | 2019-12 |
| AY278741 | SARS coronavirus Urbani | - | - |
| KY417146 | Bat SARS-like coronavirus | Rhinolophus sinicus | 2013-04-17 |
| MN996532 | Bat coronavirus RaTG13 | Chiroptera | 2013-07-24 |
Annotate the nucleotide sequences and save in a dataframe
This function iterates over a dataframe that matches the accessions in the sequences we have loaded previously. This returns a dataframe with all the annotated sequences in one table. They can then be extracted based on whatever critierion we need. Note the annotated record for each file is written to a genbank file and it’s loaded if present instead of re-running the annotation (even though the annotation only takes a couple of seconds for a virus). The core method called here is run_annotation from a package that I wrote called pathogenie. It performs a Prokka-type annotation on prokaryotic sequences and returns a dataframe and list of one or more SeqRecord objects. This function does use blast, hmmer, aragorn and prodigal but they can all be installed easily on Linux.
def annotate_files(df):
"""Annotate a set of sequences from a dataframe"""
outdir = 'annot'
res = []
for i,row in df.iterrows():
label = row.Accession
#print(row)
gbfile = os.path.join(outdir,label+'.gbk')
if os.path.exists(gbfile):
featdf = pathogenie.tools.genbank_to_dataframe(gbfile)
featdf['sequence'] = featdf.translation
else:
seq = seqrecs[label]
filename = os.path.join('temp',label+'.fasta')
SeqIO.write(seq,filename,'fasta')
featdf,recs = pathogenie.app.run_annotation(filename, threads=10, kingdom='viruses')
pathogenie.tools.recs_to_genbank(recs, gbfile)
featdf['label'] = label
featdf['host'] = row.Host
featdf['id'] = row.Species
res.append(featdf)
res = pd.concat(res)
return res
annot = annotate_files(subset)
We can then see which proteins are present in the dataframe by looking at the ‘product’ field. You can see that the same names are present in all annotations.
annot['product'].value_counts()
hypothetical protein 15
Replicase polyprotein 1a 5
Protein 3a 5
Spike glycoprotein 5
Membrane protein 5
Replicase polyprotein 1ab 5
Protein 7a 5
Nucleoprotein 5
Get a protein sequence of interest across all the annotations
get_similar_sequences simply takes the product name and finds it’s sequence in each annotation from the dataframe produced previously. It’s crude because it relies on the product name being the same. A better method would be to find all orthologs at some level of sequence similarity.
def get_similar_sequences(protname, annot):
"""Extract similar sequences by name from a set of annotations"""
seqs = []
for i,df in annot.groupby('label'):
s = df[df['product']==protname]
if len(s)==0:
continue
s = s.iloc[0]
#print (s)
seq = SeqRecord(Seq(s.sequence),id=s.label)#,description=s.host)
seqs.append(seq)
return seqs
seqs = get_similar_sequences('Spike glycoprotein', annot)
Alternative method
Obviously it would probably be easier in this case to search the relevant protein sequence on genbank and use blastp to get it’s closest hits. Then just download and align them. This assumes the protein sequences you need are in the database though. The method I have shown can be re-used for any sequences without repeating those manual steps every time.
Align the sequences
We then just align those sequences with Clustal and show the alignment in the notebook interactively using another package called pybioviz. This works inside a Jupyter notebook, but there are lots of alignment viewers available. If you scroll along the sequence you will see the RBD and polybasic cleavage site in MN908947.
aln = pathogenie.tools.clustal_alignment(seqs=seqs)
p = plotters.plot_sequence_alignment(aln)
show(p)
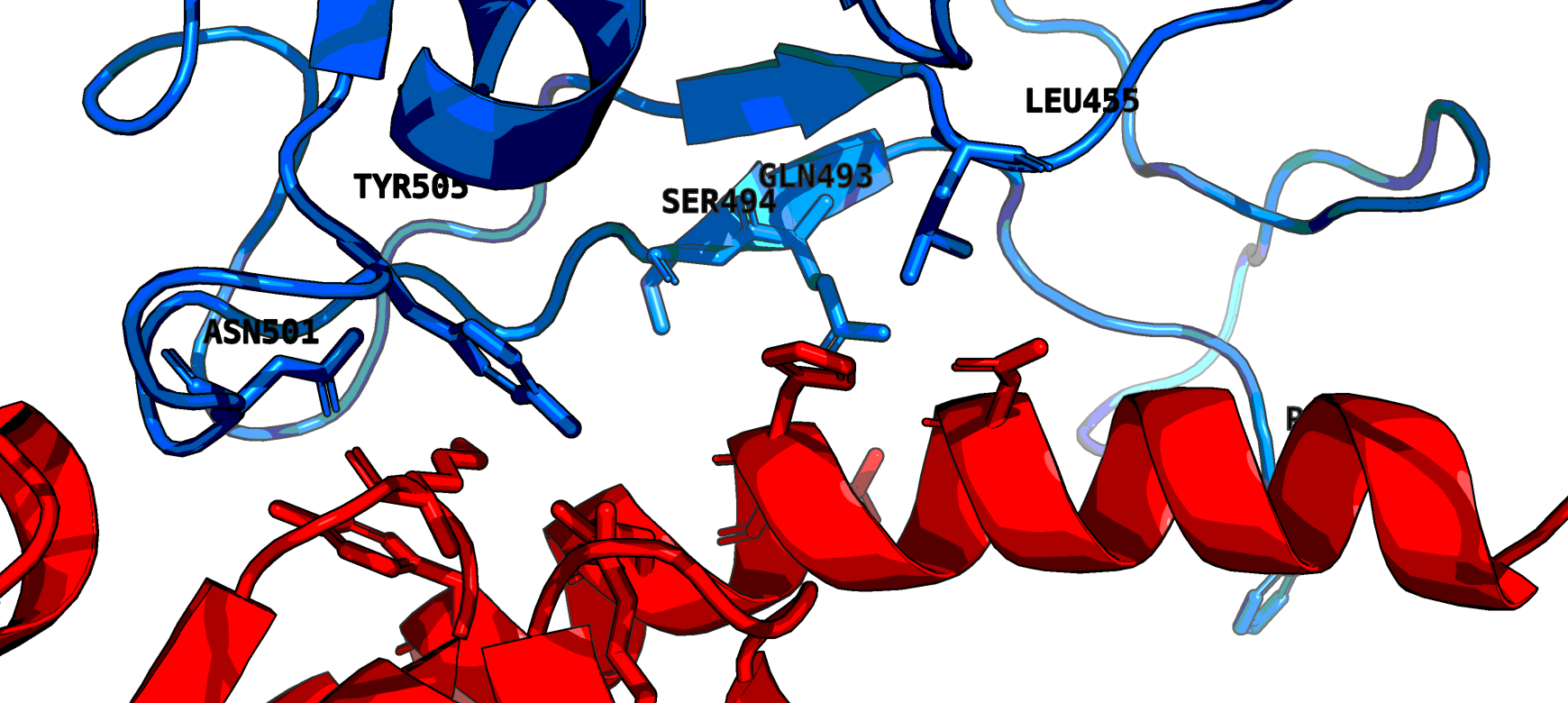
Structural view of receptor binding domain
Let’s say we want to take a look at the effect of these sequence changes on the structure of the protein. You can view models of the virus protein structures on SWISS-MODEL here: https://swissmodel.expasy.org/repository/species/2697049. They are based on the closest proteome and associated known structures. The particular model of interest here is the heterodimer Spike protein complexed with the human ACE2 receptor. Here we show a basic example of how to view the binding domain with PyMol. We can use Python to load and set up a scene focused on the interacting residues. The mutated residues are L455, F486, Q493, S494, N501, and Y505 using the coordinates of the model structure. The method find_interacting_residues selects the six key residues on chain C, the Spike protein and finds all residues in the receptor (chain D) within 4Å of any of these. This gives us a simple picture of the interaction. More in depth study of the structure is beyond the scope of this article.
def find_interacting_residues():
"""Find set of residues"""
from pymol import stored
vals = {}
residues = [455,486,493,494,501,505]
offset=3
for p in residues:
sel1 = '(c. C and (donor or acceptor) and resi %s)' %p
cmd.select('near','c. D within 4 of %s' %sel1)
cmd.show('stick','near')
cmd.color('red', 'near')
stored.lst=[]
cmd.iterate('near',"stored.lst.append((chain,resi,resn,name))")
#print (stored.lst)
for r in stored.lst:
cmd.show('stick','resi %s' %r[1])
return
cmd.reinitialize()
cmd.load('model_spike.pdb')
cmd.orient()
cmd.turn('z', 50)
cmd.bg_color('white')
cmd.select('rbd', '(chain C and resi 455+486+493+494+501+505)')
cmd.zoom('rbd')
cmd.color('marine','chain C')
cmd.color('red','chain D')
cmd.show('sticks', 'rbd')
cmd.label('rbd and n. c' , 'resn+resi')
cmd.set('label_position', (1,2,3))
find_interacting_residues()
cmd.set('ray_trace_mode',3)
cmd.png('sarscov2_spike_ace.png', width=1200,dpi=150)
Image(filename='sarscov2_spike_ace.png')
cmd.save('sarscov2_spike_ace.pse')
References
- Wan, Y., Shang, J., Graham, R., Baric, R. S. & Li, F. Receptor recognition by novel coronavirus from Wuhan: An analysis based on decade-long structural studies of SARS. J. Virol. (2020) doi:10.1128/JVI.00127-20.
- Hoffmann, M. et al., The novel coronavirus 2019 (2019-nCoV) uses the SARS-coronavirus receptor ACE2 and the cellular protease TMPRSS2 for entry into target cells. bioRxiv 2020.01.31.929042 (2020) doi:10.1101/2020.01.31.929042.
- Song W, Gui M, Wang X, Xiang Y (2018) Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog 14(8): e1007236. https://doi.org/10.1371/journal.ppat.1007236