Run bcftools mpileup in parallel with Python
BAM files and pileups
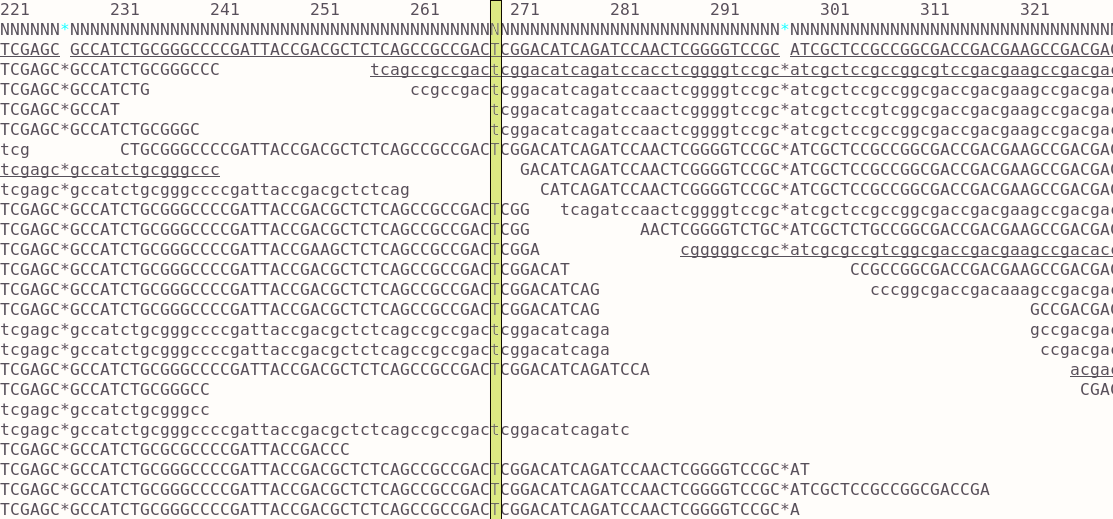
When aligning short reads to a reference genome, the result is kept as a bam file. Each position in the reference is covered by the set of reads aligning to that region. Often the purpose of doing this is to call variants of the sample against the reference. To do this you first need to essentially ‘pile up’ all the bases from any reads covering a position (a single column in the image) and check them against the reference genotype. In most cases they will be the same since you’re dealing with very similar genomes. Where there is an SNP there will be some difference and the majority might show a mutation from A->T for example. These changes might be due to sequence errors but if you have good coverage you can rule those out. Quality scores are assigned to the calls and used to filter them. These basic steps can be done using bcftools using a few commands: essentially pileup and then call variants.
There are various software tools to do this, one is bcftools. The two steps are as follows:
bcftools mpileup -O b -o <out> -f <ref> <bam files>
bcftools call --ploidy 1 -m -v -o {b} <out>
The pileup (bcftools mpileup) step is time consuming but is not multithreaded. The solution is to split the genome by region or chromosome and then join the results together. This can be done with gnu-parallel in Linux. Another method is to run via Python with subprocess to call the commands. The method shown here works for microbial genomes with one chromosome, so the genome is split into regions and each run is called in parallel. We then use bcftools concat to join the resulting bcf files. The idea is shown below.

This requires you install bcftools. On Linux this is easily done through the package manager.
Imports
import sys,os,subprocess
import numpy as np
from Bio import SeqIO
from pyfaidx import Fasta
import multiprocessing as mp
Utility methods
def get_chrom(filename):
rec = list(SeqIO.parse(filename, 'fasta'))[0]
return rec.id
def get_fasta_length(filename):
"""Get length of reference sequence"""
refseq = Fasta(filename)
key = list(refseq.keys())[0]
l = len(refseq[key])
return l
Code
This method implements mpileup in parallel using the Python multiprocessing module. You need to create a worker method that will be called by pool.apply_async. In our case wach worker will call bcftools mpileup for a specific region. The regions should not overlap. We take the entire genome length and split the coordates into equally sized blocks, depending on the number of threads we want to use. Each worker outputs it’s own bcf file. These can then be joined when finished using bcftools concat. Note this works for any number of bam files, but the samples should be the same for all workers. You could likely also used the Threading module for this, but I am less familiar with that.
def worker(region,out,bam_files):
"""Run bcftools for a single region."""
cmd = 'bcftools mpileup -r {reg} -O b -o {o} -f {r} {b}'.format(r=ref, reg=region, b=bam_files, o=out)
#print (cmd)
subprocess.check_output(cmd, shell=True)
cmd = 'bcftools index {o}'.format(o=out)
subprocess.check_output(cmd, shell=True)
return
def mpileup_parallel(bam_files, ref, outpath, threads=4, callback=None):
"""Run mpileup in parallel over multiple regions, then concat vcf files.
Assumes alignment to a bacterial reference with a single chromosome."""
bam_files = ' '.join(bam_files)
rawbcf = os.path.join(outpath,'raw.bcf')
tmpdir = 'tmp'
chr = get_chrom(ref)
length = get_fasta_length(ref)
#find regions
bsize = int(length/(threads-1))
x = np.linspace(1,length,threads,dtype=int)
blocks=[]
for i in range(len(x)):
if i < len(x)-1:
blocks.append((x[i],x[i+1]-1))
pool = mp.Pool(threads)
outfiles = []
for start,end in blocks:
print (start, end)
region = '{c}:{s}-{e}'.format(c=chr,s=start,e=end)
out = '{o}/{s}.bcf'.format(o=tmpdir,s=start)
f = pool.apply_async(worker, [region,out,bam_files])
outfiles.append(out)
pool.close()
pool.join()
#concat files
cmd = 'bcftools concat {i} -O b -o {o}'.format(i=' '.join(outfiles),o=rawbcf)
subprocess.check_output(cmd, shell=True)
#remove temp files
for f in outfiles:
os.remove(f)
return rawbcf
You can call the function like this. Then do the usual call command with the resulting bcf.
rawbcf = mpileup_parallel(bam_files, ref, outpath, threads=10)