Using epitopepredict for MHC binding prediction in Python
Background
The prediction of peptide binding to MHC molecules is the main way to do T cell epitope prediction. This is used in computational design of vaccines or immunidiagnostics that rely on using immunogenic peptides to evoke cell mediated immune responses. Typically binding predictors are based on training models with experimental binding affinity measurements made using known peptide sequences. Most of this experimental data is for human alleles and hence the binding prediction algorithms are mainly useful for human applications. Though there have been improvements in recent years to cover animal MHC alleles.
Many binding prediction tools have web interfaces but there is no way to conveniently compare the results from these predictors. It is difficult to integrate results from multiple sequences and alleles. The results are usually in a format specfic to the web tool. Command line tools offer better control, though may be harder to use. epitopepredict is a Python package that has a command line interface and API for accessing some common prediction algorithms and treating the results in a common format.
This post covers some of the basics in using epitopepredict from within Python. For those who want to use the command line tool instead you can look at the documentation.
Install the Python package
sudo pip install epitopepredict
Create a predictor and run for some random peptides
Let us create a predictor object using the ‘basemhc1’ method which is built-in and run it on some random peptides.
import pandas as pd
import epitopepredict as ep
P = ep.get_predictor('basicmhc1')
from epitopepredict import peptutils
#get some random peptides, returns a list
seqs = peptutils.create_random_sequences(10)
#run predictions
res = P.predict_peptides(seqs, alleles='HLA-A*01:01')
The predict_peptides command returns a dataframe like the example below. The dataframe is sorted by allele and rank.
peptide pos log50k score name allele rank
0 VIVRYMQHY 8 0.2376 3823.80 temp HLA-A*01:01 1.0
1 QPEIFIWMI 1 0.2104 5132.24 temp HLA-A*01:01 2.0
2 VHCWGMGFP 5 0.1508 9780.60 temp HLA-A*01:01 3.0
3 YFEWVHGHV 6 0.0991 17112.05 temp HLA-A*01:01 4.0
4 FIGGTPFVR 7 0.0934 18200.62 temp HLA-A*01:01 5.0
What prediction tools are available
A number of binding prediction tools are wrapped by epitopepredict. You can get the list shown below and use these identifiers to create your prediction object. Only the basemhc1 and tepitope methods are built-in. You can install the others using the documentation. Unless there is a good reason we recommend mhcflurry for class-I prediction and netmhciipan for class-II. If using alleles that are not well covered by mhcflurry use netmhcpan.
print base.predictors
['basicmhc1', 'tepitope', 'netmhciipan', 'netmhcpan', 'mhcflurry', 'mhcnuggets', 'iedbmhc1', 'iedbmhc2']
Which alleles to use?
The get_preset_alleles lets you pick some useful preset alleles (see docs for list).
m2alleles = base.get_preset_alleles('mhc2_supertypes')
print (m2alleles)
['HLA-DRB1*01:01', 'HLA-DRB1*03:01', 'HLA-DRB1*04:01', 'HLA-DRB1*07:01', 'HLA-DRB1*08:01', 'HLA-DRB1*11:01', 'HLA-DRB1*13:01', 'HLA-DRB1*15:01']
Running in parallel
Most predictors support using multiprocessing to run predictions in parallel if you want to speed things up. You can call either predict_peptides or predict_sequences with the cpus=n argument where n is the number of threads:
res = P.predict_peptides(seqs, alleles='HLA-A*01:01', cpus=4)
Predict for a set of proteins
You can read in protein sequences from a fasta or genbank file and predict the binders in the same way using predict_sequences. If you pass a path argument the results will be saved to a folder instead of being kept in memory. Useful for large numbers of proteins.
prots = ep.genbank_to_dataframe('MTB-H37Rv.gb',cds=True)
prots[:5]
P = ep.get_predictor('tepitope')
#predict the first 20 proteins
mb_binders = P.predict_sequences(prots[:20], alleles=m2alleles, cpus=4)
Promiscuous binders
Binding promiscuity generally means peptides that are binders in multiple alleles. The default method is to use allele-specific percentile based cutoffs.
#assumes the predictor object has data loaded into it
pb = P.promiscuous_binders(n=3)
print (pb[:3])
peptide pos name alleles core score mean median_rank
43 AILMLYTIVII 359 Rv0017c 8 ILMLYTIVI 6.32 4.120195 16.0
386 GLVMIHRLDLV 91 Rv0017c 7 LVMIHRLDL 8.10 5.093849 1.0
733 LRIMQAQLLAK 136 Rv0004 7 LRIMQAQLL 6.80 4.135143 1.0
#score based cutoff, may give different results
pb = P.promiscuous_binders(n=3, cutoff=500, cutoff_method='score')
Plot predicted binders in a sequence
#see what protein sequences we have stored in the object
print (P.get_names())
['Rv0001', 'Rv0002', 'Rv0003', 'Rv0004', 'Rv0005', 'Rv0006', 'Rv0007', 'Rv0008c', 'Rv0009', 'Rv0010c', 'Rv0011c', 'Rv0012', 'Rv0013', 'Rv0014c', 'Rv0015c', 'Rv0016c', 'Rv0017c', 'Rv0018c', 'Rv0019c', 'Rv0020c']
ax = plotting.plot_tracks([P],name='Rv0011c',cutoff=.95,n=2)
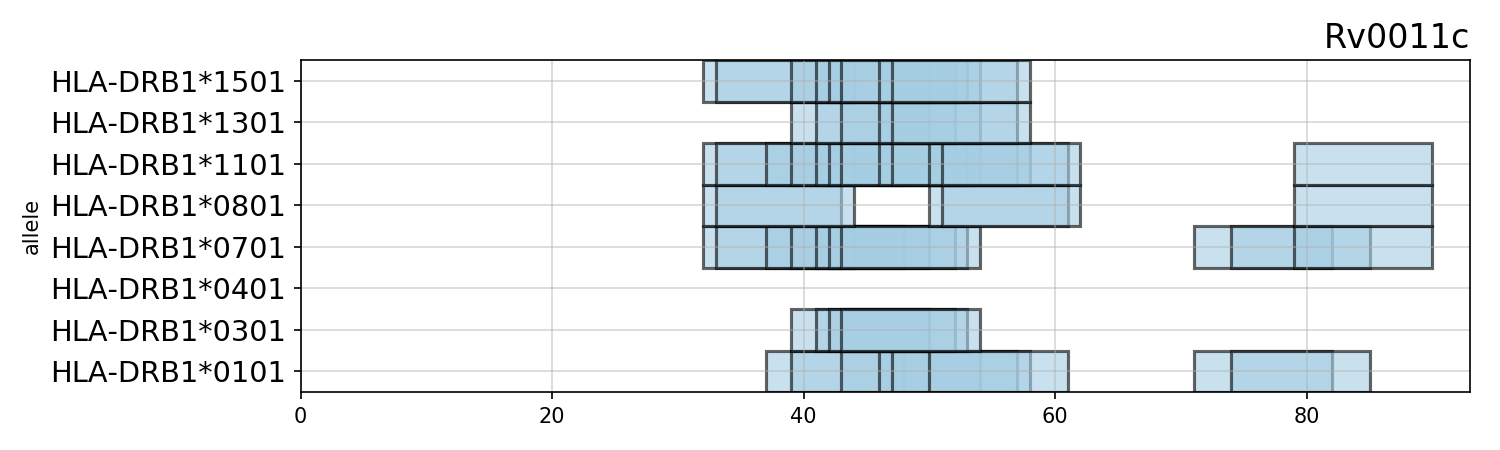
#plots a heatmap style view colored by ranks
ax = plotting.plot_binder_map(P,name='Rv0011c',cutoff=10)

Jupyter notebooks
- These examples and others are viewable in Jupyter notebooks here.
- You can also open the notebooks on binder and interact with them using this link
Links
References
- V. Jurtz, S. Paul, M. Andreatta, P. Marcatili, B. Peters, and M. Nielsen, “NetMHCpan-4.0: Improved Peptide–MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data,” J. Immunol., vol. 199, no. 9, 2017.
- T. J. O’Donnell, A. Rubinsteyn, M. Bonsack, A. B. Riemer, U. Laserson, and J. Hammerbacher, “MHCflurry: Open-Source Class I MHC Binding Affinity Prediction,” Cell Syst., vol. 7, no. 1, p. 129–132.e4, 2018.