An individual based model of farm pathogen spread with Python/Mesa
Background
Computer-based disease spread models are frequently used in veterinary science to simulate disease spread. So called Agent based (ABM) or individual based models simulate each ‘agent’ in a network of contacts. Each agent could be an animal or other entity like a herd with certain behaviours. For example the agents can randomly contact each other and transmit disease. Running such a simulation with many heterogeneous entities over many steps produces emergent complex behaviour that is meant to approximate real life disease spread. Comparing the output to real world data may allow estimation of some of the input parameters such as the transmission rates between species for example. We should always keep in mind the aphorism that ‘all models are wrong but some are useful’ in the sense that they are crude representations of the real world. Thus using them for prediction is fraught with problems in any field. But they can serve a very useful purpose in that they generate information which is otherwise difficult to obtain. They can also be useful decision-making tools by simulating surveillance or control measures.
A previous post on this blog showed how to use the Mesa library to make a agent based model for generic disease spread. Here we try to apply the same tools to make a more realistic herd pathogen model for bovine TB spread amongst herds and wildlife. It was used to generate simulated data that can be tested against a regression model. This model has not been validated against real data as yet and sensitivity testing of parameters is needed.
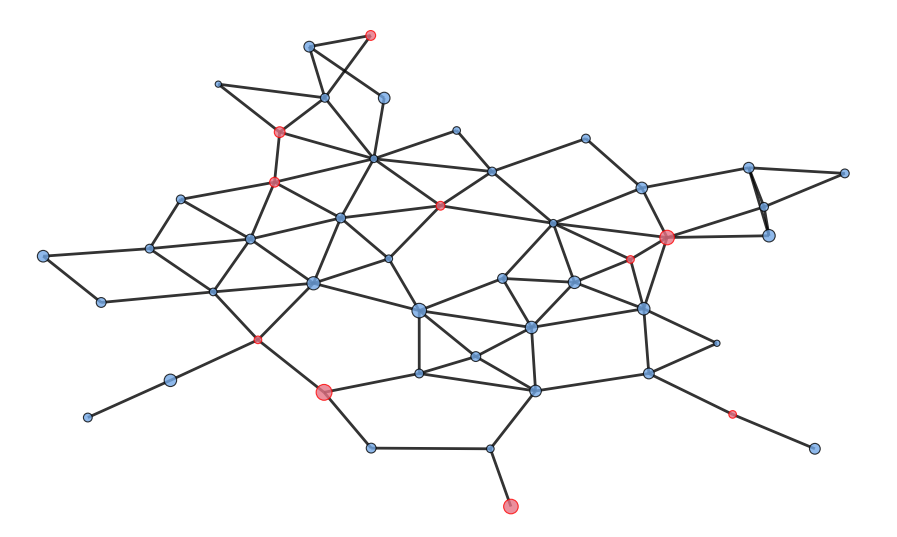
Typical network with herds represented by blue nodes and red setts.
Implementation
Mesa uses a NetworkGrid class that stores represents each node (usually an agent). This is constructed from a networkx graph that represents herd connectivity. This would reflect spatial distance of farms and land parcels for example. In this implementation each node in the grid (and corresponding network graph) actually represents a herd or badger sett. Herd and Sett objects are defined that contain a group of connected animals, also defined is an Animal class that can be a Cow or Badger. These entities are designed to have different behaviour in terms of contact, life span etc. Each animal can be in one of three defined states, Susceptible, Latent or Infected. In the latent state animals don’t transmit. This state could also represent exposed/immune animals that never transmit onwards before death. At each step cows/badgers at the same node can contact each other and randomly transmit the infection, if present, with a given probability. Animals in herds with connecting nodes can also have contacts with a lower probability. In this way if we start the model with a few infected animals, this will transmit around the network. Animals will die after a certain time if infected or naturally. The animal is then replaced to keep the population stable.
This model was designed to produce spatio-temporal and genetic simulated data. The positions in the network can be seen to represent centroids on a map. To simulate genetic relatedness we assign a strain for each infected animal. This can mutate randomly at a given rate by assigning an artificial DNA sequence and altering it by a single nucleotide during the infection process. If we start the model by setting n animals as infected with a predefined ‘strain’ we can then extract the sequences of all circulating and/or past strains and make a phylogeny from this alignment.
Parameters
- mean_stay_time - mean time in a herd
- mean_inf_time - mean infection time
- mean_latency_time - mean latency time when not infectious
- cctrans - cow-cow transmission prob
- bctrans - badger-cow transmission prob
- infected_start - how many cows to infect at start
- mean_inf_time - mean infection time length before death
- mean_stay_time - mean time on farm
- seq_length - sequence length for simulating strains/mutations
- allow_moves - whether to allow movements between farms
Movements
In real life agriculture cattle are traded between farms which means an infected animal could appear in a herd far away and spread that strain locally. By default animals in the model don’t move from the farm until they die. Movement can be added though it brings an extra complexity to the spread of individual strains.
Usage
The model is implemeented in a package called bTBabm available on github here. If you want to try it out, the easiest way is to install with pip:
pip install -e git+https://github.com/dmnfarrell/btbabm.git#egg=btbabm
We run the model as follows:
from btbam import models, utils
model = models.FarmPathogenModel(100,2000,5,graph_seed=5,seq_length=100,allow_moves=False)
for i in range(2000):
model.step()
We can visualise the network at it’s current state using the following function with typical outputs shown below.
utils.plot_grid(model,ax=ax,pos=model.pos,colorby='strain',ns='num_infected')
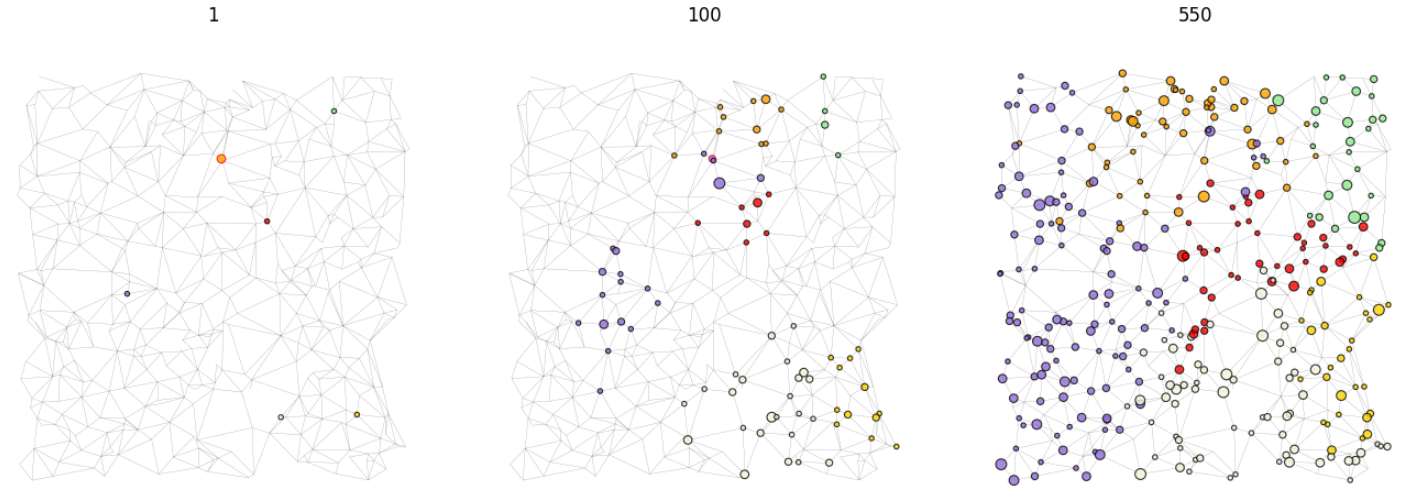
Grid plot at various steps of the simulation.
There is also a panel dashboard used for visualising the network and model outputs as it steps. In the example below you can see the phylogeny changing as the model is run.
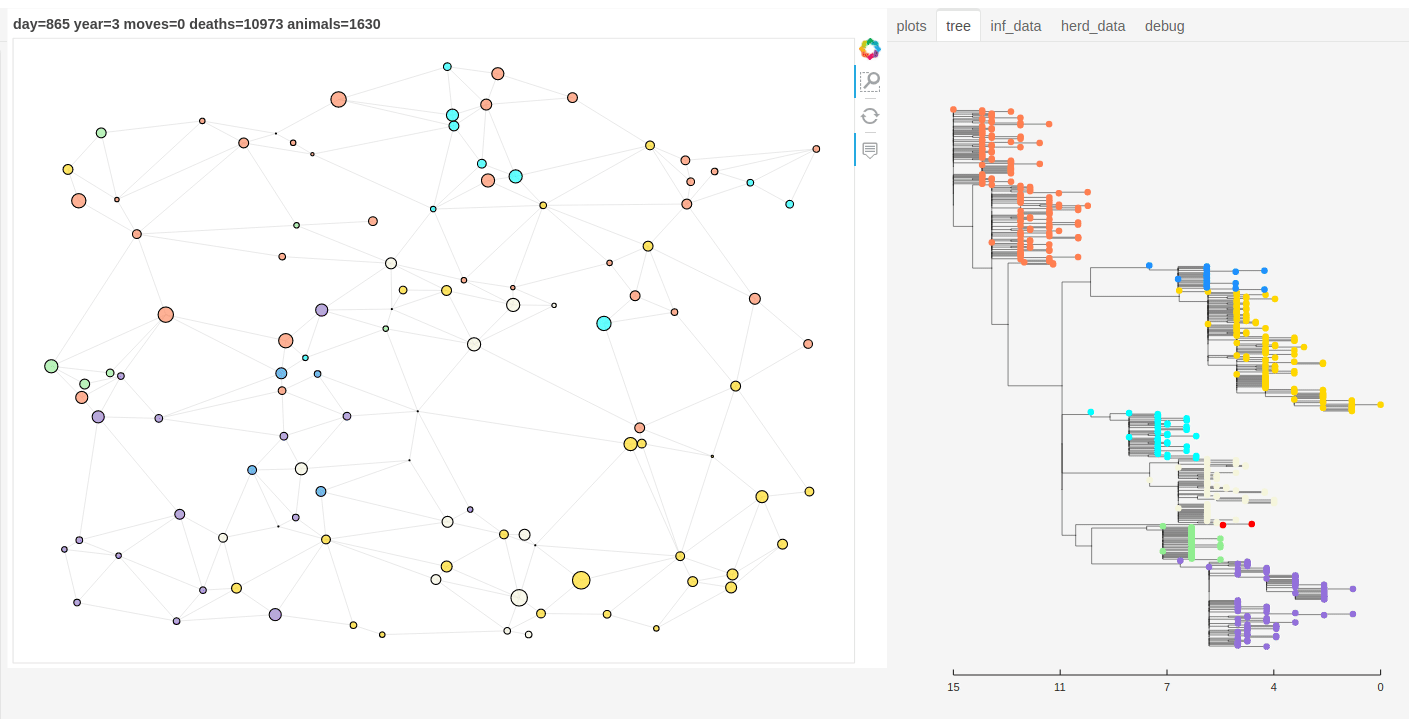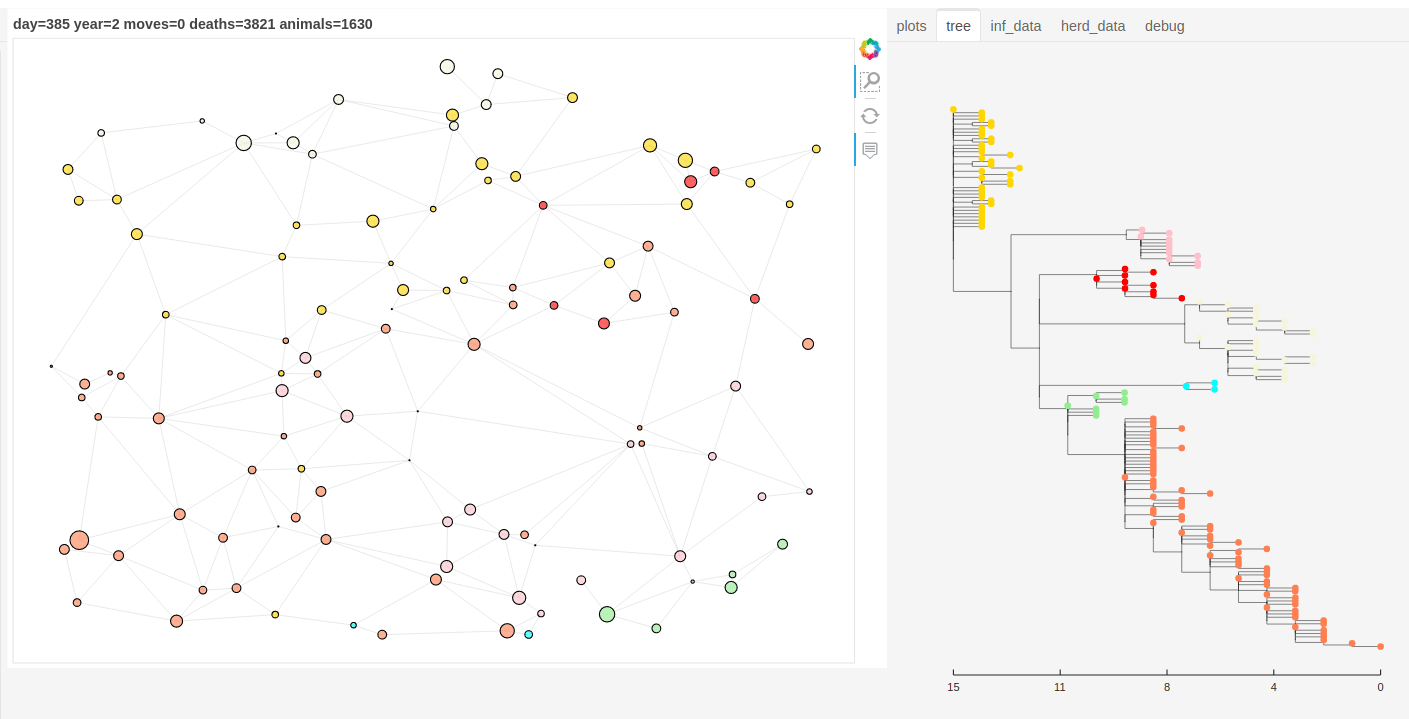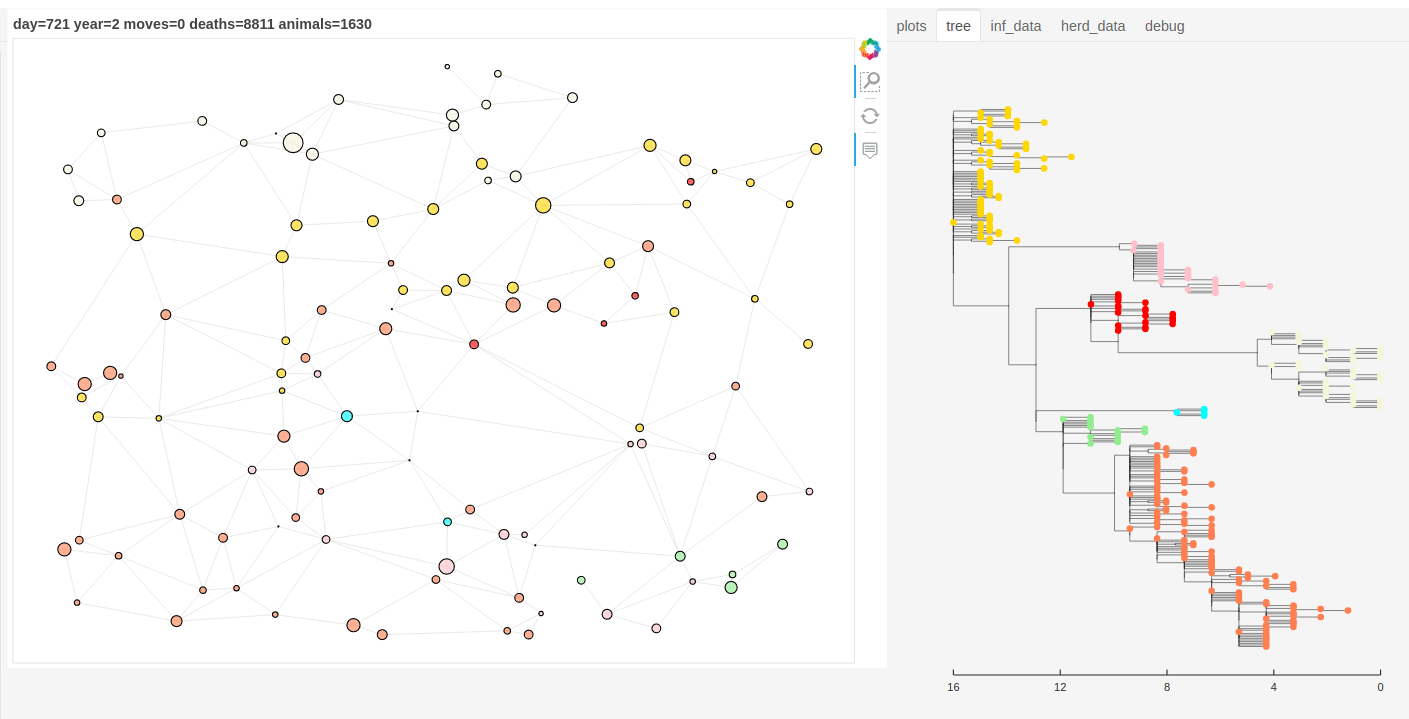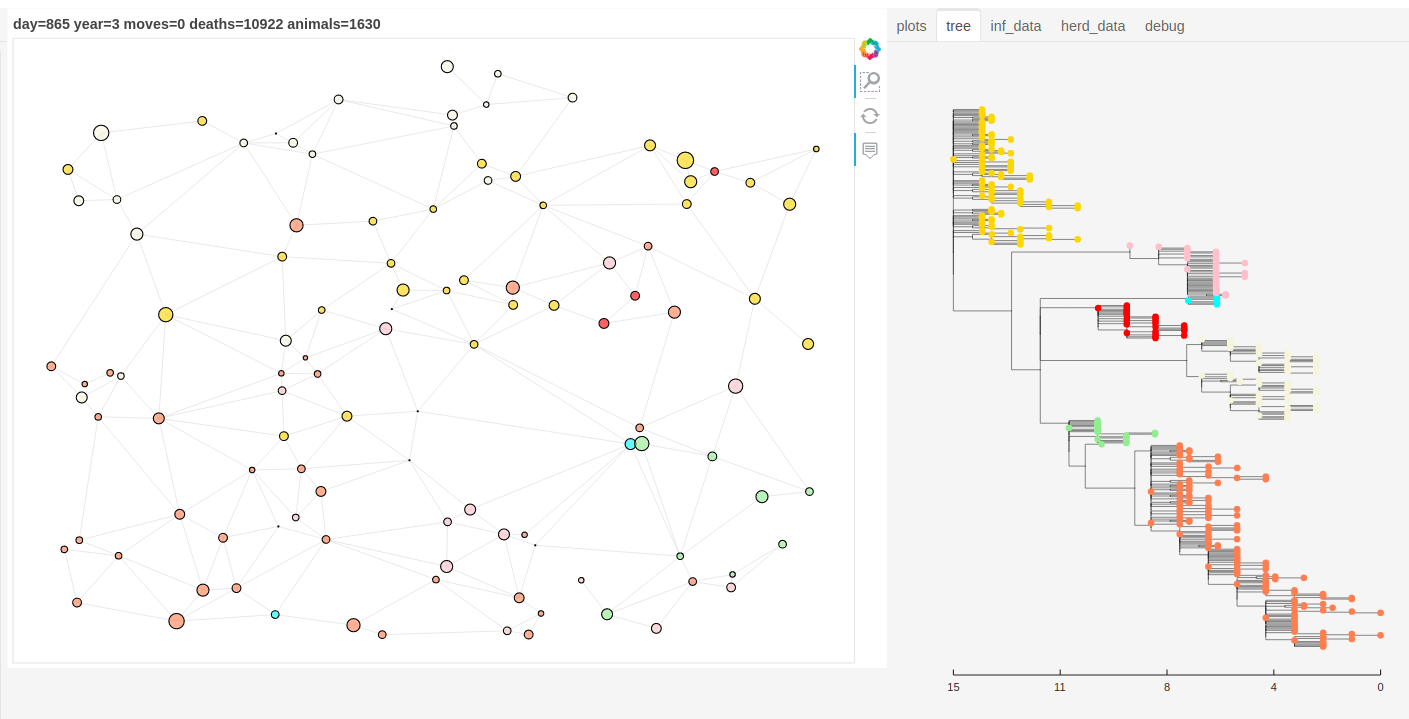
Model network and tree example.
Getting outputs
To get some outputs we can use the following functions. They all return pandas dataframes.
df = model.get_column_data() #states over all steps
df.plot()
model.get_herds_data() #will give us the per herd data at the current step
model.get_animal_data() #get the per herd data at the current step
model.make_phylogeny(removed=True,redundant=False) #make a phylogeny from the strains
get_column_data() produces this plot of each state over time. Note how the model has reached an equilibrium.
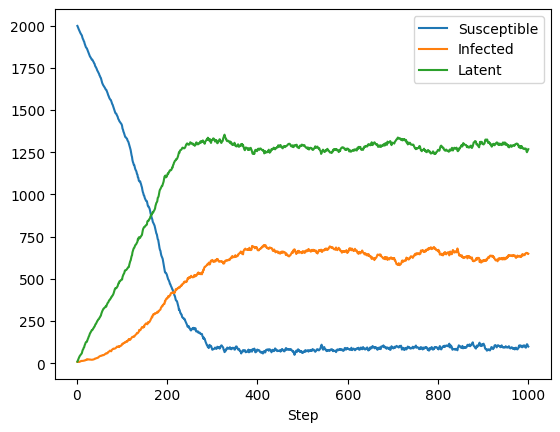
get_column_data() gets the state values at each step.
Summary
This is an initial attempt at making a model of bTB pathogen spread that was made to create simulated location and genetic distances for testing purposes. The code may be of use to those wishing to implement their own Python agent based models for similar diseases. The model here has not been tested except in the sense of code validation.
Links
References
- A Practical Introduction to Mechanistic Modeling of Disease Transmission in Veterinary Science. Kirkeby et al. Front. Vet. Sci., 26 January 2021 link
- Individual-based model for the control of Bovine Viral Diarrhea spread in livestock trade networks. Bassett et al. link
- Representing Tuberculosis Transmission with Complex Contagion: An Agent-Based Simulation Modeling Approach. Zwick et al. Medical Decision Making April 27, 2021. link