Genome annotation with BLAST, Prodigal and Biopython
Background
Genome annotation is the process of identifying the coding and non-coding features in a set of genomic DNA sequences. Usually the sequences will come from a draft assembly in the form of contigs. The features are labelled and recorded in various file formats such as genbank or gff files. They can be displayed as tracks in genome browsers. One such tool is Prokka. This is designed for bacterial and viral annotation and is written in Perl. The Prokka method takes a hierarchial approach to make it fast. A small, core set of well characterized proteins from UniProt are first searched using BLAST+. Then a series of slower but more sensitive HMM databases are searched using HMMER3. A program called Prodigal is first used to determine all the open reading frames (ORFs) in the input sequence(s).
A simplified version of the Prokka method is as follows:
- run prodigal
- blast output protein seqs
- select best hits for each unique location in the genome
- make genbank from results
The following Python code shows a method to carry out the steps above on an input fasta file. It is a bare bones method only and uses a single file of UniProt Sequences as it’s search set for BLAST. This code uses the core sequence file produced by Prokka from the set of curated UniProt bacterial proteins, UniProtKB. The fasta sequences have the following header format and this is used later to parse the protein product and gene names when we make the genbank file.
>Q92AT0 2.4.1.333~~~~~~1,2-beta-oligoglucan phosphorylase~~~COG3459
This code requires Biopython, Pandas, the ncbi-blast+ tools and Prodigal to be installed. Note that some of the methods used are called from a module tools. That code is not included for brevity. Those methods are available in this repository
import sys,os,subprocess
from Bio.SeqRecord import SeqRecord
from Bio.Seq import Seq
from Bio import SeqIO
from Bio.SeqFeature import SeqFeature, FeatureLocation
from Bio.Alphabet import generic_dna
import pandas as pd
#some helper functions
def prodigal(infile):
"""Run prodigal"""
name = os.path.splitext(infile)[0]
cmd = 'prodigal -i {n}.fa -a {n}.faa -f gff -o {n}.gff -p single'.format(n=name)
subprocess.check_output(cmd, shell=True)
resfile = name+'.faa'
return resfile
def get_prodigal_coords(x):
#get coords from prodigal fasta header
s = re.split('\#|\s',x.replace(' ',''))
coords = [int(i) for i in s[1:4]]
return pd.Series(coords)
def prokka_header_info(x):
#extract infor from prokka header
s = re.split('~~~',x)
return pd.Series(s)
All the main steps are inside a single function below which might be a bit unwieldy for some tastes.
def annotate_contigs(infile, outfile=None, **kwargs):
"""
Annotate nucelotide sequences (usually a draft assembly with contigs)
using prodigal and blast to prokka seqs. Writes a genbank file to the
same folder.
Args:
infile: input fasta file
outfile: output genbank
returns:
a list of SeqRecords with the features
"""
#run prodigal
resfile = prodigal(infile)
#get target seqs
seqs = list(SeqIO.parse(resfile,'fasta'))
#make blast db of prokka proteins
fetch_sequence_from_url('sprot', path=prokkadbdir)
dbname = os.path.join(prokkadbdir,'sprot.fa')
tools.make_blast_database(dbname, dbtype='prot')
print ('blasting ORFS to uniprot sequences')
bl = tools.blast_sequences(dbname, seqs, maxseqs=1, evalue=1e-3,
cmd='blastp', show_cmd=True, **kwargs)
bl[['protein_id','gene','product','cog']] = bl.stitle.apply(prokka_header_info,1)
cols = ['qseqid','sseqid','pident','sstart','send','protein_id','gene','product']
bl = bl.sort_values(['qseqid','pident'], ascending=False).drop_duplicates(['qseqid'])[cols]
#read input file seqs
contigs = SeqIO.to_dict(SeqIO.parse(infile,'fasta'))
#read in prodigal fasta to dataframe
df = tools.fasta_to_dataframe(resfile)
df[['start','end','strand']] = df.description.apply(get_prodigal_coords,1)
#merge blast result with prodigal fasta file info
res = df.merge(bl,left_on='name',right_on='qseqid',how='right')
#get simple name for contig
def get_contig(x):
return ('_').join(x.split('_')[:-1])
res['contig'] = res['name'].apply(get_contig)
l=1 #counter for assigning locus tags
if outfile is None:
outfile = infile+'.gbk'
handle = open(outfile,'w+')
recs = []
#group by contig and get features for each protein found
for c,df in res.groupby('contig'):
print (c, len(df))
contig = get_contig(c)
#truncated label for writing to genbank
label = ('_').join(c.split('_')[:2])
nucseq = contigs[c].seq
rec = SeqRecord(nucseq)
rec.seq.alphabet = generic_dna
rec.id = label
rec.name = label
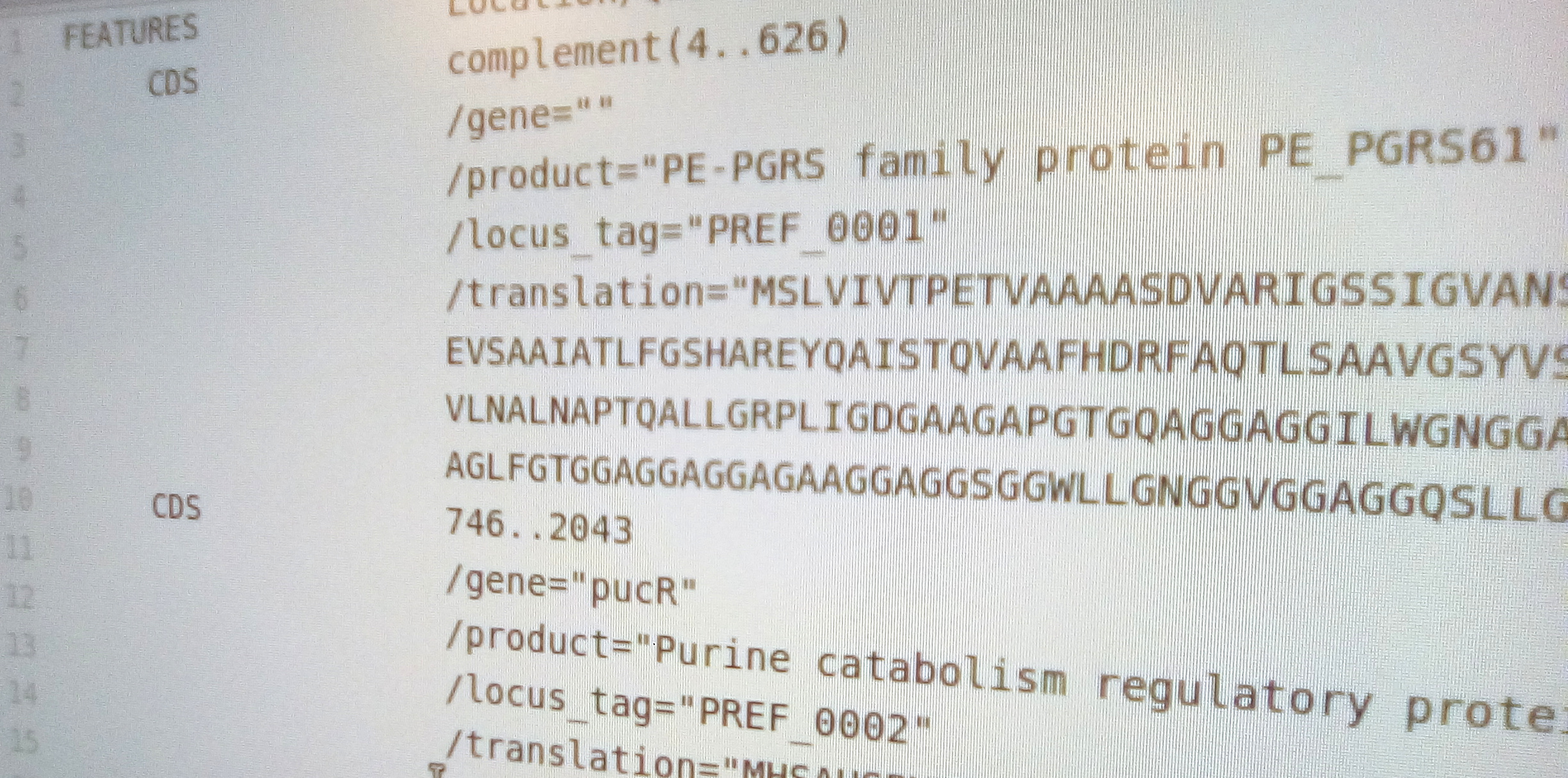
for i,row in df.iterrows():
tag = 'PREF_{l:04d}'.format(l=l)
quals = {'gene':row.gene,'product':row['product'],'locus_tag':tag,'translation':row.sequence}
feat = SeqFeature(FeatureLocation(row.start,row.end,row.strand), strand=row.strand,
type="CDS", qualifiers=quals)
rec.features.append(feat)
l+=1
#print(rec.format("gb"))
SeqIO.write(rec, handle, "genbank")
recs.append(rec)
handle.close()
return recs
We then call the function as follows. It will write the results to a basic genbank file.
annotate_contigs('myfile.fasta')
This code is a naive version of the kind of genome annotation done in Prokka. I have not included the more sensitive detection steps and any customisation. Hopefully it may be a useful starting point for writing custom annotation routines. This code is utilised in a Python package I made for resistance gene prediction called pyamrfinder.