Bacterial SNP detection with nanopore vs. illumina sequencing
Background
Previously we looked at the sensitivity of SNP detection in M.bovis Illumina by running two independent sequencing runs for 16 M.bovis isolates. Libraries were prepared separately from the same cultures. Illumina sequencing was performed on two different platforms, a NextSeq 500 with 150 paired end reads and a MiSeq with ~250-300 paired end reads. The data was then analysed in SNiPgenie by running all samples together and then retrieving the SNP distance matrix. From this we could compare the SNP distance between each corresponding pair, which should be ideally zero.
Here we do the same thing for three of the samples but now also comparing to nanopore sequencing runs. Oxford nanopore (ONT) sequencing produces long reads but with higher error rates than short read platforms. The technology is relatively new but has progressed rapidly, with new flow cell chemistry and software to call bases being introduced.
Basecalling
Nanopore sequencers produce small changes in electrical current detected across the pores as nucleotides pass through. Thus the output is raw signal data which must be converted into sequence or ‘basecalled’. ONT provides software called guppy for this purpose. The process is slow and the GPU accelerated version of the software is an order of magnitude faster than running on CPU only. Guppy GPU can be installed on Ubuntu Linux fairly easily but we had to use a slightly roundabout approach to get this working on Ubuntu 21.04 using the instructions provided by Miles Benton here. The software is developed rapidly, so this may have changed by the time this article is read.
Guppy settings
There are some useful posts on GPU settings for base calling such as this one. It is also recommended to consult the nanopore community forum if you have access.
There are many command line options for guppy, the function of which is not at first clear. To optimise the calling for your hardware you will want to utilise the GPU memory and processor threads you have without crashing the program. This can be done by trial and error.
--num_callers: The number of callers, dictates the maximum number of CPU threads used. There is always one CPU support thread per GPU caller.--chunks_per_runner: The maximum number of chunks which can be submitted to a single neural network runner before it starts computation. Increasing this figure will increase GPU basecalling performance when it is enabled.--gpu_runners_per_device: The number of neural network runners to create per CUDA device. Increasing this may improve performance on GPUs with a large number of compute cores, but will increase GPU memory use.
The general rule for memory use:
runners x chunks_per_runner x chunk_size < 100000 * [max GPU memory in GB]
It is recommended to use nvtop and htop to view memory and CPU usage. Too many gpu_runners_per_device will fill GPU memory.
Testing
We first tested the basecalling with the phage lambda genome provided by ONT. The flow cell type used was FLO-MIN106 with a single Minion device and SQK-LSK109 sequencing kit. We used a Ryzen 3900X CPU and Nvidia RTX 3060 with 12GB RAM for base calling. The guppy version was 5.0.16+b9fcd7b5b. After testing with the fast model we found the settings below seemed reasonable.
| setting | value |
|---|---|
| model_file | template_r9.4.1_450bps_hac.jsn |
| chunk_size | 2000 |
| cpu_threads_per_caller | 20 |
| num_callers | 20 |
| chunks_per_runner | 512 |
| gpu_runners_per_device | 5 |
The fast model took about 76 seconds to run our phage lambda data and the HAC (accurate) model about 10 minutes, so it’s a lot slower. The HAC model with above settings took 44 minutes to process a set of M.bovis reads.
Results
We sequenced three M.bovis samples individually (no multiplexing) and so had quite good depth per sample as follows:
TB20-3762 154.73
TB19-3825 130.61
TB20-3738 624.09
Variants were called with SNiPgenie along with the two sets of previous illumina runs. So we could then compare the SNP distance of each triplicate in the following cluster map that shows all vs all distances:
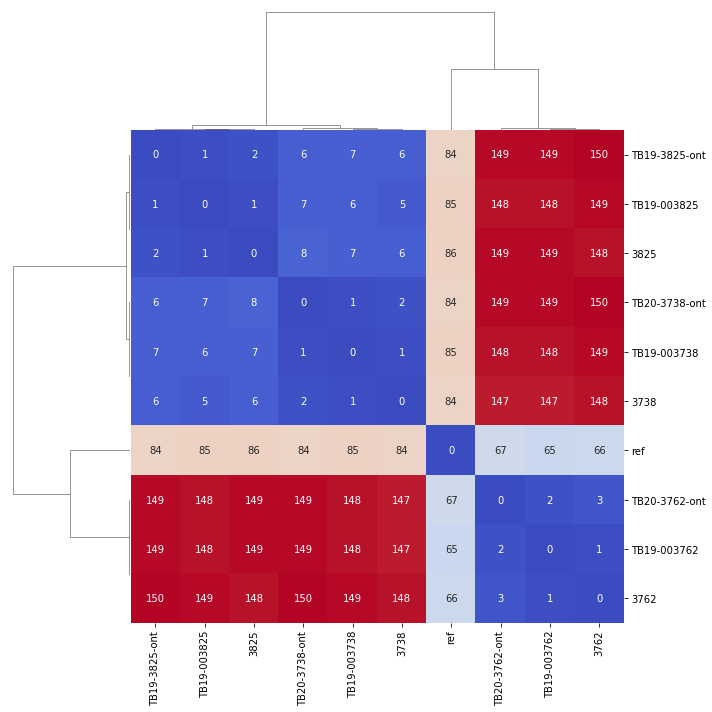
Differences in SNPs called between three duplicate runs, one of which is from nanopore sequencing.
Obviously the triplicates cluster together with 0-2 SNPs between them except for one sample which had 3. One site called differently for nanopore is shown below and is a result of a heterozygous SNP in the other data not being called. This illustrates that we can certainly use nanopore sequencing for this purpose, though we do not know how lower depths will affect results. To make such a method cost effective would probably require multiplexing of samples which will limit read depth.
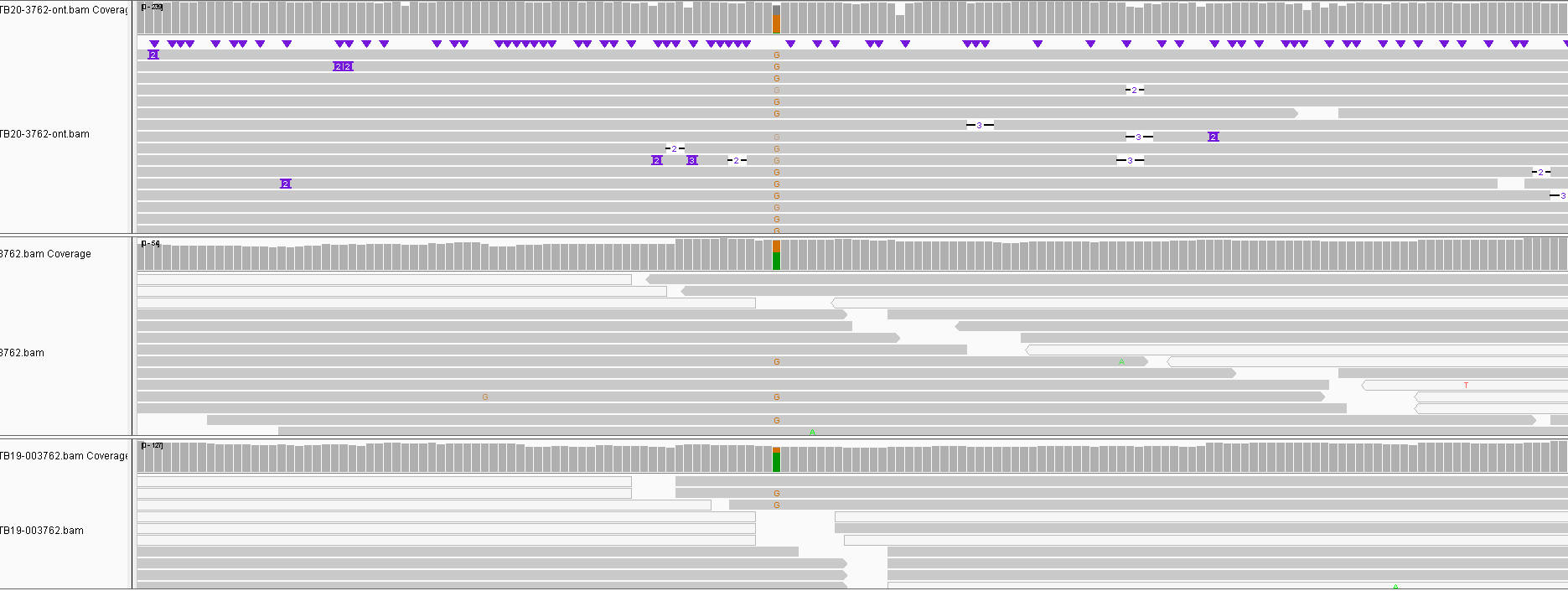
A location called differently for nanopore reads.
Notes
Altering some of the guppy settings such as num_callers and cpu_threads_per_caller didn’t seem to make much difference. Increasing gpu_runners_per_device from 1 to 2 had the biggest effect, beyond this the speed increases were much less, but more memory was used. I may not be using the program correctly! Note that versions of guppy>=5.0 use more memory so GPUs with at least 6GB RAM are possibly recommended for applications like adaptive sampling that require rapid basecalling.