M. bovis spoligotyping from WGS reads
Background
Spoligotyping (spacer oligonucleotide typing) is a widely used genotyping method for M. tb (Mycobacterium tuberculosis species), which exploits the genetic diversity in the direct repeat (DR) locus in Mtb genome. Each DR region consists of several copies of the 36 bp DR sequence, which are interspersed with 34 bp to 41 bp non-repetitive spacers. A set of 43 unique spacer sequences is used to classify Mtb strains based on their presence or absence. This a molecular method traditionally conducted using a PCR-based or other method. Whole genome sequence is a far more sensitive method of phylogenetic identification of strains and makes other typing methods redundant if you have the whole sequence. It may be useful however to relate the sequence back to the spoligotype in some circumstances. It is not hard to calculate the spoligotype by analysis of the raw sequence reads (or an assembly).
Method
The method is simple and consists of blasting the spacer sequences against a database made from the raw reads. The reads can be assembled or concatentated together first but the method will work regardless. The results are parsed and hits per spacer counted. A threshold is used to determine if a spacer is present or not. Low coverage hits and those with less than 1 or 2 mismatches should also be removed first. The leaves a 43 digit presence/absence string whihc is a binary code with 1 denoting the presence and 0 denoting the absence for each spacer. This can be translated into octal or hexadecimal code and looked up in a database of known types that correspond to that code. This is basically the same method used by the SpoTyping tool. SpoTyping uses a threshold of 5 hits by default. The value my vary according to the input reads but should be at least 2 to account for spurious hits.
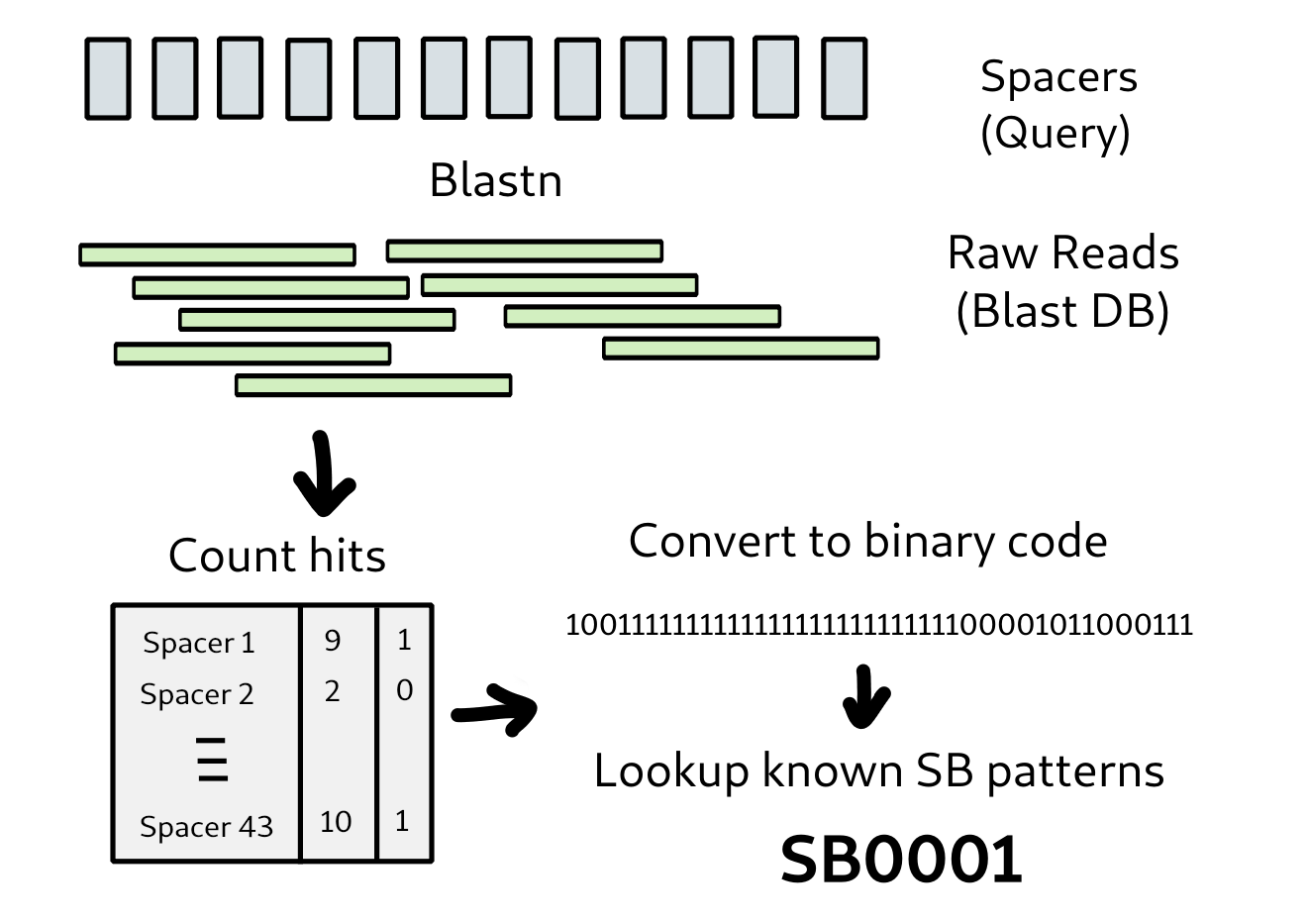
Scheme for detecting spoligotype from short reads.
Code
DR spacers are put into a fasta file with a number for each according to the order that will determine the SB number binary code. You can download this file here. The file looks like this, each sequence is 25 nucleotides long:
>1
ATAGAGGGTCGCCGGCTCTGGATCA
>2
CCTCATGCTTGGGCGACAGCTTTTG
>3
CCGTGCTTCCAGTGATCGCCTTCTA
>4
ACGTCATACGCCGACCAATCATCAG
>5
TTTTCTGACCACTTGTGCGGGATTA
We then just make the blast database from the reads by translating the fastq file into fast format and using makeblastdb. The following method does this and the retrieval of hits. It puts the results into a pandas DataFrame and filters them. They are then aggregated to get hits per spacer. This is converted to the binary code. The methods make_blast_database and blast_fasta are imported from the tools module in the snpgenie package. They can be copied from the repository if needed separately. This method uses the reads direct without assembly or concatentation. It limits to the first 500000 reads for efficiency but this could be changed. If you have paired end reads it will probably work using one of the files.
from snpgenie import tools
def get_spoligotype(filename, reads_limit=500000, threshold=2):
"""Get spoligotype from reads"""
ref = 'dr_spacers.fa'
#convert reads to fasta
tools.fastq_to_fasta(filename, 'temp.fa', reads_limit)
#make blast db from reads
tools.make_blast_database('temp.fa')
#blast spacers to db
bl = tools.blast_fasta('temp.fa', ref, evalue=0.1,
maxseqs=100000, show_cmd=False)
#filter hits
bl=bl[(bl.qcovs>95) & (bl.mismatch<2)]
#group resulting table to get hits per spacer sequence
x = bl.groupby('qseqid').agg({'pident':np.size}).reset_index()
#filter
x = x[x.pident>=threshold]
found = list(x.qseqid)
s=[]
#convert hits to binary code
for i in range(1,44):
if i in found:
s.append('1')
else:
s.append('0')
s =''.join(s)
return s
We can then look up the binary code in the Mbovis.org database using a table downloaded form the website. There may be a more comprehensive source. The table looks like this:
SB,binary,octal,hexadecimal
SB0001,1001111111111111111111111111000010110001111,477777777413071,4F-7F-7F-7F-0B-0F
SB0002,1001111111111011111111111111000010110001111,477757777413071,4F-7E-7F-7F-0B-0F
SB0009,1101101000001100111111111111111111110100000,664063777777200,6D-03-1F-7F-FF-20
SB0040,1101111101111110111111111111111110001100000,676773777770600,6F-5F-5F-7F-F8-60
SB0054,1100101000001110111111111111111111111100000,624073777777600,65-03-5F-7F-FF-60
SB0058,1111111111110111111111111111111100001000111,777737777760431,7F-7D-7F-7F-F0-47
SB0063,1111111111111111110111001111111100001001111,777777347760471,7F-7F-7B-4F-F0-4F
..
def get_sb_number(binary_str):
"""Get SB number from binary pattern usinf database reference"""
df = pd.read_csv('Mbovis.org_db.csv')
x = df[df['binary'] == binary_str]
if len(x) == 0:
return
else:
return x.iloc[0].SB
Use as follows:
b = get_spoligotype('test.fastq')
get_sb_number(b)
This method appears to work on test data with known types but hasn’t been rigorously benchmarked. It’s not likely to be 100% reliable and results should be checked for errors. You will notice that a single missed hit will produce the wrong result entirely.