A simple genome browser with Qt and dna_features_viewer
Background
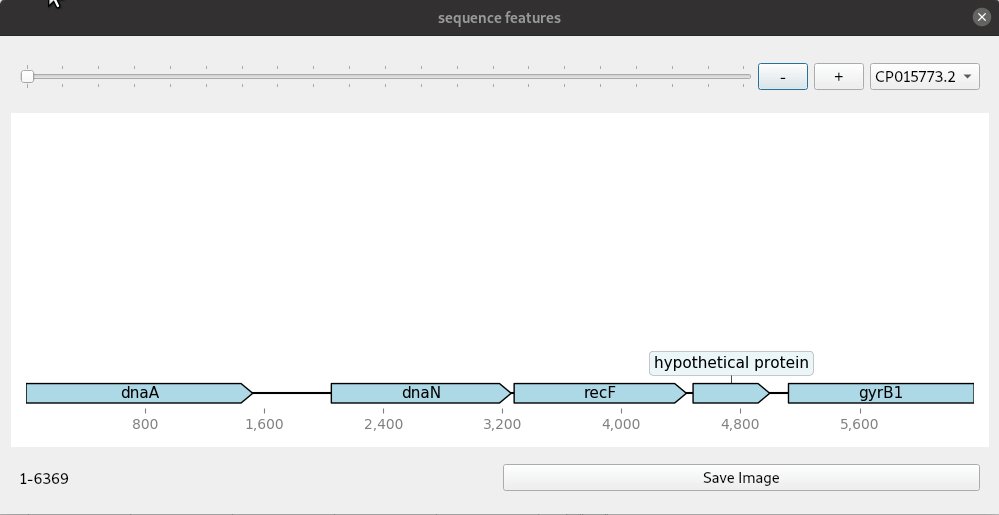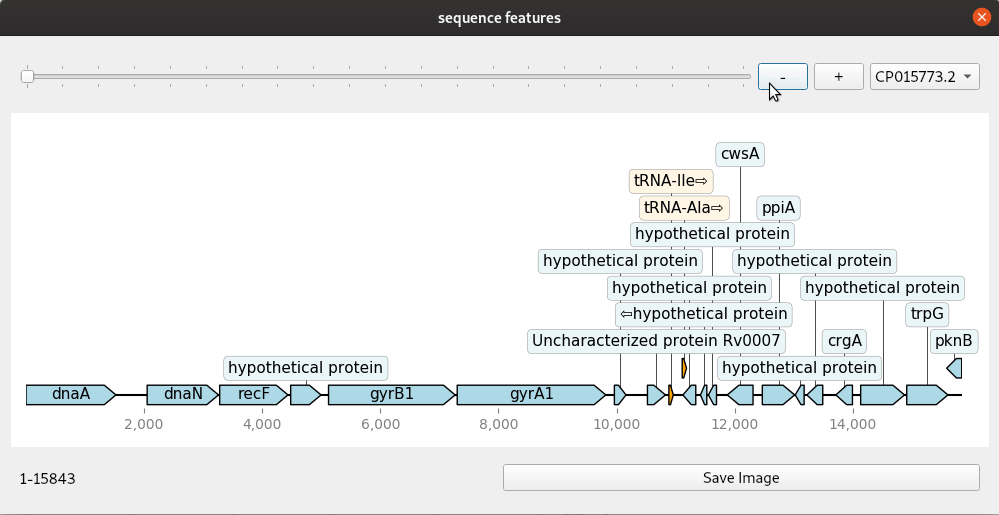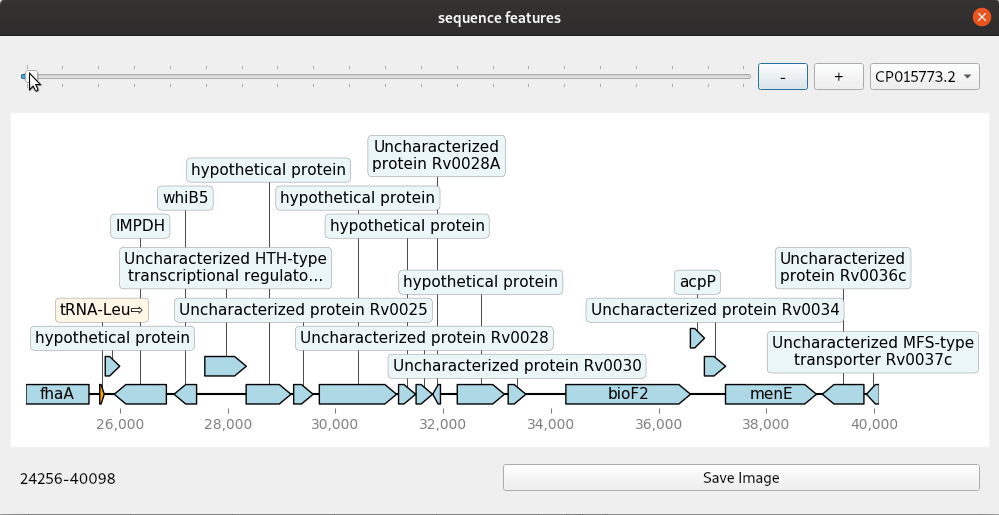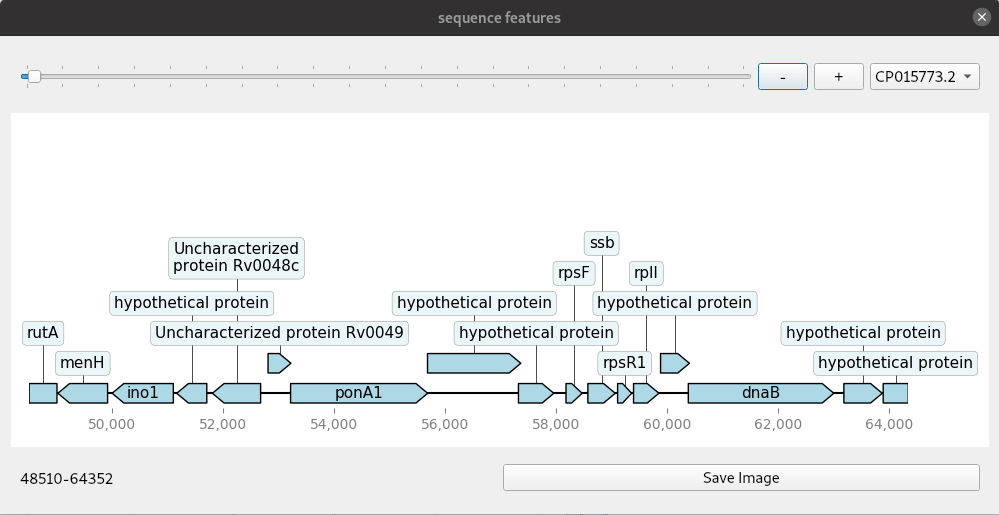
Genome browsers are regularly used to view genomic annotations (features) in web browsers or desktop programs. These are mostly run on a web server and customisable to some extent with the ability to add and remove tracks. Increasingly, users will want more customised browsers because there are many more different kinds of datasets than ever. Building your own custom genome viewer is not trivial though. Most efforts now focus on web technologies due to their portability. Desktop solutions do have some advantages if you can make use of the power of the graphical interface toolkits they provide. A simple example shown here uses dna_features_viewer, a Python package for drawing genomic features using matplotlib. Combined with the Qt toolkit we can quickly make a small application that makes a handy, though simple, genome browser. The Qt widgets are implemented with PySide2.
Imports
import sys,os
from Bio import SeqIO
from PySide2 import QtCore
from PySide2.QtWidgets import *
from PySide2.QtGui import *
import matplotlib
import pylab as plt
from dna_features_viewer import GraphicFeature, GraphicRecord
from dna_features_viewer import BiopythonTranslator
The application
The code for this mini application is given here in one block. The update method is where the drawing is done. This is connected to any changes in the slider and zoom buttons so that a redraw is done each time the co-ordinates change. The program handles genbank files with one or more contig or continuous sequence. These are selected from the drop down menu. Files are read in with Biopython and converted to a list of SeqRecord objects.
class SeqFeaturesViewer(QMainWindow):
"""Sequence records features viewer using dna_features_viewer"""
def __init__(self, genbank=None, gff=None):
QMainWindow.__init__(self)
self.setAttribute(QtCore.Qt.WA_DeleteOnClose)
self.setWindowTitle('Genomic Features Viewer')
self.setGeometry(QtCore.QRect(300, 200, 1000, 400))
self.setMinimumHeight(150)
self.main = QWidget()
self.main.setFocus()
self.setCentralWidget(self.main)
self.add_widgets()
self.color_map = {
"rep_origin": "yellow",
"CDS": "lightblue",
"regulatory": "red",
"misc_recomb": "darkblue",
"misc_feature": "lightgreen",
"tRNA": "lightred"
}
if genbank != None:
self.load_genbank(genbank)
elif gff !=None:
self.load_gff(gff)
return
def load_genbank(self, filename):
"""Load a genbank file"""
recs = list(SeqIO.parse(filename, 'gb'))
self.load_records(recs)
self.update()
return
def load_gff(self, filename):
"""Load a gff file"""
from BCBio import GFF
in_handle = open(filename,'r')
recs = list(GFF.parse(in_handle))
self.load_records(recs)
self.update()
return
def add_widgets(self):
"""Add widgets"""
l = QVBoxLayout(self.main)
self.main.setLayout(l)
val=0
navpanel = QWidget()
navpanel.setMaximumHeight(60)
l.addWidget(navpanel)
bl = QHBoxLayout(navpanel)
slider = QSlider(QtCore.Qt.Horizontal)
slider.setTickPosition(slider.TicksBothSides)
slider.setTickInterval(1000)
slider.setPageStep(200)
slider.setValue(1)
slider.valueChanged.connect(self.value_changed)
self.slider = slider
bl.addWidget(slider)
zoomoutbtn = QPushButton('-')
zoomoutbtn.setMaximumWidth(50)
bl.addWidget(zoomoutbtn)
zoomoutbtn.clicked.connect(self.zoom_out)
zoominbtn = QPushButton('+')
zoominbtn.setMaximumWidth(50)
bl.addWidget(zoominbtn)
zoominbtn.clicked.connect(self.zoom_in)
self.recselect = QComboBox()
self.recselect.currentIndexChanged.connect(self.update_record)
bl.addWidget(self.recselect)
from matplotlib.backends.backend_qt4agg import FigureCanvasQTAgg as FigureCanvas
import matplotlib.pyplot as plt
fig,ax = plt.subplots(1,1,figsize=(15,2))
self.canvas = FigureCanvas(fig)
l.addWidget(self.canvas)
self.ax = ax
bottom = QWidget()
bottom.setMaximumHeight(50)
l.addWidget(bottom)
bl2 = QHBoxLayout(bottom)
self.loclbl = QLabel('')
bl2.addWidget(self.loclbl)
savebtn = QPushButton('Save Image')
savebtn.clicked.connect(self.save_image)
bl2.addWidget(savebtn)
return
def load_records(self, recs):
"""Load list of SeqRecord objects"""
from Bio import SeqIO
self.records = SeqIO.to_dict(recs)
recnames = list(self.records.keys())
self.rec = self.records[recnames[0]]
length = len(self.rec.seq)
self.recselect.addItems(recnames)
self.recselect.setStyleSheet("QComboBox { combobox-popup: 0; }");
self.recselect.setMaxVisibleItems(10)
sl = self.slider
sl.setMinimum(1)
sl.setMaximum(length)
sl.setTickInterval(length/20)
return
def update_record(self, recname=None):
"""Update record"""
recname = self.recselect.currentText()
self.rec = self.records[recname]
length = len(self.rec.seq)
sl = self.slider
sl.setMinimum(1)
sl.setMaximum(length)
sl.setTickInterval(length/20)
self.update()
return
def value_changed(self):
"""Callback for widgets"""
length = len(self.rec.seq)
r = self.view_range
start = int(self.slider.value())
end = int(start+r)
if end > length:
end=length
self.update(start, end)
return
def zoom_in(self):
"""Zoom in"""
length = len(self.rec.seq)
fac = 1.2
r = int(self.view_range/fac)
start = int(self.slider.value())
end = start + r
if end > length:
end=length
self.update(start, end)
return
def zoom_out(self):
"""Zoom out"""
length = len(self.rec.seq)
fac = 1.2
r = int(self.view_range*fac)
start = int(self.slider.value())
end = start + r
if end > length:
end=length
self.update(start, end)
return
def update(self, start=1, end=2000):
"""Plot the features"""
ax=self.ax
ax.clear()
if start<0:
start=0
if end == 0:
end = start+1000
if end-start > 100000:
end = start+100000
#print (start, end)
rec = self.rec
translator = BiopythonTranslator(
features_filters=(lambda f: f.type not in ["gene", "source"],),
features_properties=lambda f: {"color": self.color_map.get(f.type, "white")},
)
graphic_record = translator.translate_record(rec)
cropped_record = graphic_record.crop((start, end))
cropped_record.plot( strand_in_label_threshold=7, ax=ax)
if end-start < 150:
cropped_record.plot_sequence(ax=ax, location=(start,end))
cropped_record.plot_translation(ax=ax, location=(start,end),fontdict={'weight': 'bold'})
plt.tight_layout()
self.canvas.draw()
self.view_range = end-start
self.loclbl.setText(str(start)+'-'+str(end))
return
def save_image(self):
filters = "png files (*.png);;svg files (*.svg);;jpg files (*.jpg);;All files (*.*)"
filename, _ = QFileDialog.getSaveFileName(self,"Save Figure",
"",filters)
if not filename:
return
self.ax.figure.savefig(filename, bbox_inches='tight')
return
def main():
"Run the application"
import sys, os
from argparse import ArgumentParser
parser = ArgumentParser(description='Genomic Features Viewer')
parser.add_argument("-f", "--genbank", dest="genbank",default=None,
help="input genbank file", metavar="FILE")
parser.add_argument("-g", "--gff", dest="gff",default=None,
help="input gff file", metavar="FILE")
args = vars(parser.parse_args())
app = QApplication(sys.argv)
aw = SeqFeaturesViewer(**args)
aw.show()
app.exec_()
if __name__ == '__main__':
main()