Rapid Average Nucleotide Identity calculation with FastANI
Background
The Average Nucleotide Identity (ANI) is a convenient way to express the similarity of two microbial genomes in a single number. It’s the mean nucleotide identity of orthologous gene pairs shared between two genomes. This value is used to delineate species. A typical percentage threshold for species boundary is 95% ANI. It can be calculated by aligning the matching segments and finding the average identity. The alignment can be done quickly with MUMmer (ANIm method) but there are other methods too.
Say you want to compare many genomes together to make a distance matrix and species tree. You could have hundreds of samples and you need to perform an all vs. all comparison. For increasing numbers of genomes the number of pairwise comparisons goes up dramatically. If n is the number of samples, then n*(n-1)/2 comparisons must be done. So even if your method only takes 10 seconds per comparison, for 200 genomes you will have to do 19,900 comparisons and that would take 55 hours!
FastANI
FastANI is a program for fast alignment-free computation of whole-genome Average Nucleotide Identity (ANI). FastANI supports complete and draft genome assemblies. It avoids explicit sequence alignments altogether and uses k-mer sampling. This is much faster and therefore scales better. It is also still quite accurate. You can run the program as follows. The reference and query contain lists of all the files to compare and are the same for all vs. all comparisons.
fastANI --ql reference.txt --rl query.txt -o fastani.out -t 10
FastANI can be parallelized for multi-core processors. It’s done by splitting the reference genomes in several equal-size parts and searching query genome(s) against each part of the reference database independently.
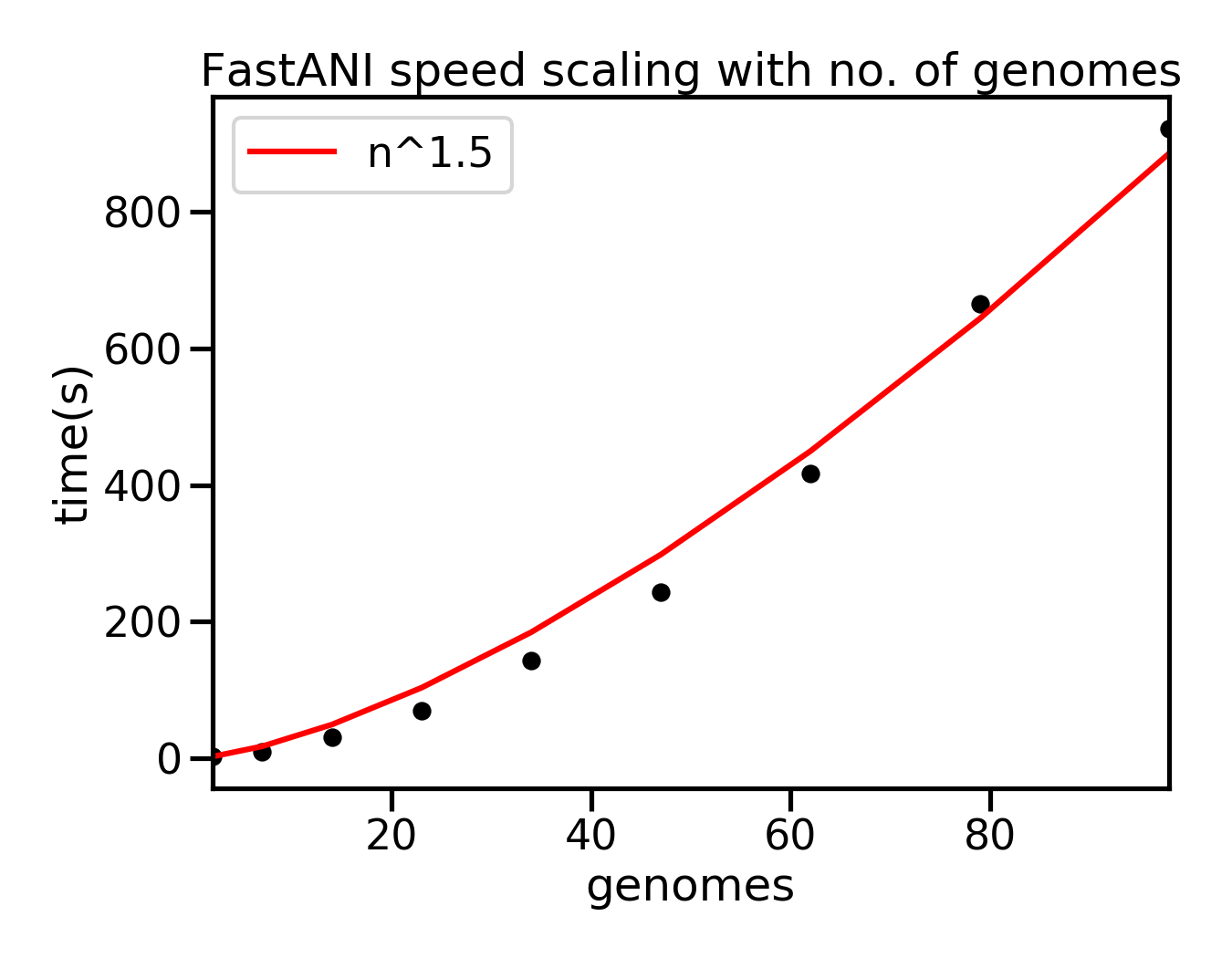
Example of scaling of FastANI with the number of genomes is shown below. Bacterial genome assemblies were used. Each run was executed in parallel with 10 threads on a Ryzen 2600X cpu. In this case the time increased approximately with no. genomes1.5. This is not a thorough benchmark but is certainly much faster than the ANIm method.
Python code
The Python code used to perform this test is given below for reference.
import os, glob, time, subprocess, shutil
def run_test(n):
#names are extracted from a dataframe here and used to make query
#and reference lists
names = list(asm.Assembly_nover)[:n]
l=[]
for f in glob.glob('assemblies/*.fa.gz'):
n=os.path.basename(f).split('.')[0]
if n in names:
l.append(f)
with open('query.txt', 'w') as infile:
infile.write('\n'.join(l))
shutil.copyfile('query.txt','reference.txt')
#run
cmd = 'fastANI --ql reference.txt --rl query.txt -o fastani.out -t 10'
st=time.time()
subprocess.check_output(cmd,shell=True)
t = time.time()-st
return t
times=[]
step=5
n=2
#run test with increasing increments of genomes
while n<=100:
t=run_test(n)
print (n,t)
times.append((n,t))
n+=step
step+=2
Read in FastANI result file to get a distance matrix of the identities.
def get_fastani(filename):
"""Get fastANI results into pairwise matrix"""
import re
df = pd.read_csv(filename,sep='\t',names=['query','ref','ident','x','y'])
#these two lines are only needed to extract the label from the file names
df['query'] = df['query'].apply(lambda x: re.split(r"[\./]+",x)[1])
df['ref'] = df['ref'].apply(lambda x: re.split(r"[\./]+",x)[1])
x = pd.pivot_table(df,index='query',values='ident',columns=['ref'])
return x
get_fastani('fastani.out')